Return to Treatments Available Page
UTube Presentations available:
Drug and Device Approaches to Heart Failure and Post-Heart Attack Outcome
Nutritional Approaches to Heart Failure and Post-Heart Attack Outcome
Bioenergetic Support in Heart Failure and Coronary Insufficiency
Integrative Approach to Heart Failure
For additional information please review a) the heartfixer.com website, b) our UTube heart failure-related presentations (Drug and Device Approaches, Nutritional Approaches to Heart Failure and Post-MI Outcome, and Bioenergetic Support for the Tired Heat), c) Reverse Heart Disease Now by Drs. Roberts and Sinatra, and d) The Sinatra Solution by Dr. Stephen Sinatra.
Definition: Cardiac pump dysfunction, impairing blood flow to our internal organs, with or without symptoms (fatigue, weakness, and shortness of breath), of any cause.
Causes:
·
Loss of heart muscle due to one or more heart attacks (ischemic cardiomyopathy),
with or without additional reversible pump function impairment due to coronary
insufficiency.
·
Cardiac strain on the basis of overload pathophysiology:
A. Afterload strain due to severe hypertension or aortic stenosis (narrowed
aortic valve).
B. Preload strain due to a leaky aortic or mitral valve or to excessive blood
recirculation due to a cardiac shunt lesion (hole in the heart) or dialysis
access placement.
C. Overuse strain, due to an elevated heart rate (Atrial Fib) or marked anemia.
·
Cardiomyopathy, a loss of heart muscle cells or cellular dysfunction due to:
A. An auto-immune response to viral infection or any other form of prior myocyte
damage.
B. Toxins, such as mercury, cobalt, and cadmium.
C. Chemotherapy for malignancy.
·
Bioenergetic strain, the loss of co-factors required to generate biochemical
energy (to recycle AMP and ADP back to ATP) such as Co-Enzyme Q10, carnitine,
magnesium, taurine, and AMP precursors.
· Multiple cause CHF (Congestive Heart Failure) – Irrespective of the cause, all patients with CHF will experience bioenergetic insufficiency and a secondary auto-immune attack against the heart muscle (myocardium).
Imaging: Cardiac catheterization, echo, or nuclear imaging, alone or in
combination, to grade pump dysfunction and quantitate secondary pulmonary
hypertension or valvular insufficiency.
Laboratory assessment:
·
BNP (B-Natriuretic peptide), a treatment responsive lab marker of CHF severity.
·
Kidney chemistries (many of our drug therapies for CHF strain kidney function).
·
EndoPAT Endothelial function testing (peripheral endothelial dysfunction
correlates with impaired myocardial bioenergetics).
·
Serum ferritin, to exclude iron overload (causes myocardial oxidative damage).
·
Markers of inflammatory and oxidative stress (Cleveland Heart and NutrEval
studies).
·
Sleep apnea assessment (not uncommon in our patients with CHF).
·
Provocative challenge testing to estimate soft tissue heavy metal burden.
·
Anabolic assessment (Testosterone in men and DHEA-s and IGF-1 in both genders).
Therapeutic Approach (at this point we have determined the cause(s) of your CHF,
graded its severity, and screened your for aggravating metabolic factors). If a
specific driving force has been identified, it will be addressed, in an effort
to prevent further damage. The following steps are or value irrespective of the
cause(s) of your CHF:
·
Bioenergetic support: Co-Enzyme Q10, carnitine, taurine, magnesium, and related
nutritional supports have been shown to improve pump function and reduce
symptoms, mortality, and hospitalization. Ribose assists with ATP energy
recycling, and arginine with endothelial tone. Nutritional testing will provide
us with specifics; listed below is a general nutritional guide:
¨Co-Enzyme
Q10 200-400 mg/day (aiming for a level
³
2.5).
¨Carnitor
330 mg tid or carnitine 500 mg bid (tid is three times a day and bid is twice a
day).
¨Ribose
5 grams tid
¨Taurine
1000 mg bid
¨Magnesium
glycinate 100-200 mg bid
· Fluid volume control: Loop diuretic therapy, furosemide or torsemide, every morning or every other morning. If necessary, metolazone (zaroxylyn) can be taken 30² prior to the loop diuretic to enhance its effect. These agents predictably waste potassium, magnesium, thiamine, and other B vitamins (we can monitor for this and supplement accordingly).
·
Neuroendocrine blockade – Biochemical “whips” elaborated by the kidneys, adrenal
glands, and nervous system in a maladaptive attempt to increase cardiac
performance will progressively damage the heart (a “secondary insult”) and must
be blocked.
¨Spironolactone
blocks the receptor for aldosterone, an adrenal hormone that leads to myocardial
stiffness, magnesium and potassium loss, and salt and water retention by the
kidneys.
¨Angiotensin
Converting Enzyme Inhibitors (ACEI) block the generation in the circulation and
(quinapril and ramipril only) in the heart and vascular wall of angiotensin II,
a powerful vasoconstrictive, inflammatory, and free radical generating peptide,
concomitantly inhibiting the degradation of bradykinin, a beneficial,
endothelial supporting molecule.
¨Angiotensin
Receptor Blockers (ARBs) do not block the generation of angiotensin II, but
prevent it from acting on its receptor, thus negating its activity. ARBs are
used primarily when ACEI is not tolerated (on the basis of cough).
¨Cardioselective
Beta Blockade (metoprolol or carvedilol) blocks maladaptive overstimulation of
the heart and kidneys by the adrenal and autonomic nervous system hormone
adrenaline. Nebivolol directly blocks the generation of superoxide free radical
and can also be used in CHF.
¨EntrestoÒ
combines an ARB (valsartan) with Sacubitril, an agent that blunts degradation of
BNP. While we use BNP as an index of CHF severity, the heart generates this
molecule in an attempt to help itself. Increasing BNP expression with EntrestoÒ
thus improves CHF symptoms.
·
Afterload reduction – Hydralazine dilates arteries, decreasing the work the
heart must do to pump blood forward. Hydralazine, like Nebivolol and
Allopurinol, also also lowers superoxide free radical generation (superoxide
destroys nitric oxide and inhibits cardiac performance).
·
SLGT2 inhibitors (Jardiance and Farxiga - also used to treat DM2) are of value
in heart failure.
·
Endothelial support – Peripheral endothelial dysfunction (low EndoPAT score)
correlates with a low nitric oxide to superoxide ratio within the myocardium,
with consequent impaired contractile efficiency (less heart function per oxygen
molecule utilized). Arginine and co-factors to encourage its conversion in to
nitric oxide can rebalance this biochemistry.
· Allopurinol
spares myocardial oxygen and blocks the generation of superoxide, while
concomitantly protecting the kidneys (from uric acid toxicity and oxidative
stress).
·
Immune modulation – The immune system’s response to heart failure is overzealous
and maladaptive and must be attenuated without compromising overall immune
defense function.
¨
Pentoxifylline blunts generation of TNF-alpha and the overblown Th1 immune
response that characterizes CHF and atherosclerosis, and has been shown to be
effective in CHF of any cause.
¨
Statins waste Co-Enzyme Q10 and by this mechanism are a negative, but they also
blunt intracellular free radical generation and down regulate the Th1 immune
response, a plus.
¨
Famotidine (Pepcid) blocks the myocardial histamine receptor, which in CHF is up
regulated.
¨
Berberine lowers inflammation and improves functional status and outcome in CHF.
¨
Vitamin D, Co-Q10, fish oil, and other nutritionals also have immune modulating
benefits.
¨
Weight loss helps on multiple fronts and will attenuate maladaptive immune
stimulation.
¨
A compounded immune modulator is available for my personal patients.
·
Khavinson peptides bioregulators provides cardiac, vascular, and liver growth factors and has
used extensively in Russia and Eastern Europe in the treatment of CHF.
· Ouabain (Stropanthus) improves contractile without increasing oxygen need, and thus is of value in heart failure and coronary insufficiency.
·
Device therapies utilize physics to improve cardiac biochemistry and function.
¨
Dual chamber pacing restores synchronicity to cardiac contraction in patients
with conduction delays, improving ejection fraction and cardiac performance.
¨
Implanted defibrillator placement provides a back stop with respect to severe
arrhythmia.
¨
EECP may improve functional status in some patients with CHF.
·
Botanical support:
¨
Hawthorne Berry 600 mg twice a day is a helpful add-on therapy (standard care in
Europe).
¨Terminalia
arjuna may help with CHF (commonly used in Asia).
James C. Roberts MD FACC FAARFM 3/4/2021
Drug Therapy for CHF - Neuroendocrine Blockade
The progressive deterioration in cardiovascular health experienced by the
patient with CHF, the drop in pump function and the worsening of symptoms,
is not due to the process that initially damaged the heart, but rather to
the body's maladaptive response to the initial injury, the release of
hormones and neurotransmitters that whip the heart to death. Modern
drug therapy for CHF effectively blocks these whips, giving the heart a
chance to heal. This concept, Neuroendocrine Blockade, will be
presented, followed by a discussion of the major drugs that we utilize (b-blockers,
Tissue Specific Angiotensin Converting Enzyme Inhibitors, Spironolactone,
Digoxin, Pentoxifylline, and Statins (yes - statin drugs for CHF).
EECP, MME, and pacemaker therapy for CHF are discussed elsewhere on this
site.
When the heart is damaged, due to muscle loss from a heart attack, strain on the basis of uncontrolled hypertension, valvular disease, or myocyte (the cells of the heart) damage due to heavy metal exposure, pump function will be impaired, and the flow of oxygenated blood to the internal organs will be compromised. Several of these organs will cry foul. They want more oxygenated blood; they will send signals to the heart, directing it to pump harder. The kidney will release renin, itself a vasoconstrictive, salt retaining hormone, that promotes the formation of angiotensin I, a weak vasoconstrictor, that is converted within the circulation and within the cells of the artery wall into angiotensin II, an incredibly potent vasoconstrictor and free radical generator. The adrenal gland will release aldosterone, which tells the kidney to retain sodium and fluid; aldosterone then travels to the heart where it promotes muscle thickening, stiffness, and fibrosis. The sympathetic nervous system goes into flight or fight mode, releasing adrenalin and nor-adrenalin, which increase the HR and BP, whipping the heart, aiming to increase the flow of oxygenated blood. The immune system is stimulated to generate free radicals and other inflammatory mediators. All of these compensatory mechanisms are designed to whip the heart into meeting the short term needs of the internal organs, and this all makes sense, that is if you are a caveman.
Our body's response to injury, to a health challenge, is the product of millions of years of evolution. It is designed to meet the needs of our predecessors. Our physiology has not been updated to address disease states that are new to man (and if you believe in Creation, you also would not expect our maker to update us every hundred years). We need to realize that the neuroendocrine outpouring that characterizes CHF was designed by evolution to address disease states that would compromise forward blood flow in pre-modern man. What might they be? Let's see, pre-modern man bleed a lot. He might loose 1/4th of his blood volume following an altercation with a saber tooth tiger. He would sweat off a lot of fluid during the fight. He would become dehydrated and his BP would fall and blood flow to the kidneys and adrenal gland would decrease. Here it makes sense for the kidneys and adrenal gland to release hormones that retain sodium and water. A jolt of adrenalin might make the difference between live and death. You want the heart to work harder. You want the blood vessels to constrict, as this will support the blood pressure, such that more blood can make it to the kidneys, adrenals, and the other vital internal organs. Cave men did not have penicillin. If the tiger gash in their arm got infected, the bacteria could spread into the blood stream. Bacterial sepsis would lower their BP, again compromising blood flow to the internal organs; the same compensatory measures, a neuroendocrine outpouring to restore the BP and restore blood flow to the internal organs, would come in to play. Atherosclerotic vascular disease, the process that leads to heart attack, was not present in man until the Egyptians began to cultivate wheat. Valvular heart disease probably did not exist 200 years ago (you would probably die of rheumatic fever or not live long enough to suffer from chronic rheumatic valvular disease). Hypertension probably did not exist 100 years ago, and heavy metal cardiac toxicity is a disease of modern man. Reduced forward blood flow in modern man is caused by these disease states, not by blood loss, dehydration, or sepsis, but our modern-day body responds to reduced forward blood flow as if it were caused by these conditions. The body's response, adaptive when carried out short term to address short term threats to our predecessors, is maladaptive when carried out long-term in modern man, who is experiencing impaired forward blood flow on the basis of intrinsic heart disease. Modern day drug therapy blocks these maladaptive responses, these hormones and angiochemicals that whip the heart, giving the heart a chance to recover, preventing what would otherwise be a steady deterioration into cardiac death. We've covered the theory. Now let's cover the specific drugs, their mechanisms of action, and their side-effects.
Diuretic Therapy - While technically not a components of neuroendocrine blockade, diuretic therapy is necessary to address the fluid retention that underlies many of the symptoms experienced by the patient with CHF. Shortness of breath, initially with effort, and later at rest or while reclining during sleep, is caused by fluid build-up within the lungs, due to impaired pumping function of the left heart. Ankle swelling, termed edema, is due to fluid retention behind the right heart. This fluid retention is brought on by neuroendocrine activity effecting the kidneys, but you are feeling it in your breathing, ankles, and sense of well being. Diuretic agents stimulate the kidney to excrete more sodium and water. As fluid is mobilized, your ankle swelling will resolve. The water will clear from your lungs and with this your breathing will improve. The fluid overload state of CHF also raises the filling pressures within the heart, stretching the heart muscle fibers out of shape, compromising their performance and damaging them. Fluid mobilization will decrease the chamber size of the cardiac pumping (ventricles) and storage (atria) chambers, removing this mechanical stress. We give you enough diuretic therapy to resolve your symptoms and "lower your filling pressures", but we try not to overdue it. If we overshoot, if we actually dehydrate you, well them forward blood flow to the kidneys will again be compromised, and guess what, there will be a second wave of neuroendocrine outpouring to contend with. We gauge your need for diuretic therapy, which may change over time, from your physical exam and symptomatic status, aided by several tests. Your chest X-ray (CXR) will show gross fluid retention behind your heart. Bioimpedence cardiac output testing assists us in determining whether you are "wet" or "dry", and by how much. B-type natriuretic peptide (BNP) is a protein made by the heart in response to hemodynamic stress. Your BNP level will rise when you are wet and fall when you are dry, and serves as a numerical marker of your fluid status. Sometimes I just can't be sure what your volume status is. Then I will carry out a right heart catheterization. Here a balloon-tipped catheter is inserted in the femoral vein (vein to the leg) and passed under X-ray guidance into the right heart, such that I can actually measure the filling pressure within your heart. We use these measures to dose your diuretic therapy with our best accuracy. We also need to be aware of the side-effects of diuretic therapy, outside of volume depletion due to excessive dosing. All diuretic agents waste potassium, magnesium, thiamine, and B vitamins; they go out with the urine. Thiamine and magnesium are critical to cardiac energy production, while low potassium leads to cardiac electrical instability. It is critical that we monitor your nutritional status as we treat your CHF. It is insane to give you these drugs and not correct for their nutritional downsides. It is rare for me to see a new diuretic treated CHF patient whose ionized magnesium level is not low. 80% of patients taking 80 mg/day of Lasix will be thiamine deficient. Thiamine deficiency will not show up in your routine lab studies but Thiamine supplementation will increase your ejection fraction by about 5% and improve your symptomatic status. Providing you with the nutritional supplements that are wasted by the diuretic drugs necessary to resolve your fluid retention is important in the Integrative Cardiology approach to CHF.
Hydrochlorothiazide (HCTZ) is a relatively weak diuretic, useful in hypertension control but typically not strong enough to meet the needs of the patient with CHF. More powerful "loop" diuretics, such as Furosemide (trade name Lasix), Torsemide (Demadex), and Bumetanide (Bumex) are typically used. Metolazone (Zaroxylyn) by itself is a fairly weak diuretic, but servers to prime the kidneys fior the action of the loop agents. I may ask you to take Metolazone (known to patients as the "kicker") 2.5 to 5 mg., 30 minutes before your morning Furosemide dose, when Furosemide alone is not enough. Amongst these agents Torsemide and Bumetanide are the most effective, as their adsorption from the GI tract is not compromised should the GI tract itself become congested with fluid overload (we call this passive congestion), but their cost is 10 times that of Furosemide, which has a long history of effective use, making it our first line diuretic agent.
b-adrenergic Blocking Agents (Beta Blockers) - This approach to CHF is at first thought counter-intuitive, but not in the context of neuroendocrine blockade. Adrenaline and nor-adrenaline, the chemical mediators (neurotransmitters) elaborated by the sympathetic nervous system, elicit the fight or flight response, our body's threat response. They affect the cardiovascular system by increasing HR, BP, cardiac pumping function, and forward blood flow. To trigger these effects, first these blood-borne agents must bind to adrenergic receptors on the outer membranes of cardiovascular cells. These receptors are one of three types: alpha (a), beta-1 (b1), or beta-2 (b2). Drugs are available that block one or more of these receptors. The alpha receptor mediates peripheral vasoconstriction, raising blood pressure and increasing the workload of the heart (the heart must work harder to pump blood into this high resistance circuit). Alpha blockade would thus seem to be a logical approach to CHF. As an intern and medical resident, I recall using Minipress, an alpha blocker, in the treatment of CHF. The idea was to block the alpha receptor, dilate the peripheral arteries, and thus "unload" the heart. The concept is valid, but we soon learned that if we dilate the blood vessels to lower the BP, the heart would compensate (inappropriately) with an increase in HR. The increased HR would increase the heart muscle's demand for oxygenated blood, and if the patient had blocked arteries (usually the case) then coronary insufficiency could develop. Minipress helped with the the CHF, but as a down-side produced coronary insufficiency, at times leading to heart attack. Alpha-blockade as a mono-therapy has thus been abandoned. The beta-2 receptor mediates contractility of the heart, the ability of the heart to squeeze with vigor when needed, which we don't want to block. Beta-2 receptors also open up our airways. The beta-1 receptors mediate the deleterious cardiac effects of adrenaline and nor-adrenaline. The beta-1 receptor mediates apoptosis (basically the premature suicide of the cardiac cell under strain). This is the receptor that we wish to block. The beta-blockers available 20 years ago (Propranolol, trade name Inderal, Nadolol, trade name Corgard, and Atenolol, trade name Tenormin) block both the beta-1 and beta-2 receptors. They could block adrenergically mediated apoptosis, but they could also compromise pump function and bring on asthma attacks in pre-disposed individuals. Metoprolol (trade names Lopressor or Toprol) blocks only the beta-1 receptor. Lopressor and Toprol block apoptosis without worsening pump function or precipitating bronchospasm. These agents, when added to standard diuretic therapy, have been shown not only to decrease symptoms and improve the functional status of CHF patients, they also decrease mortality - you live better and you live longer. "Cardio-specific" beta-1 blockade with Metoprolol thus makes sense in CHF. Carvedilol (trade name Coreg) was introduced 5 years ago. Coreg blocks both beta receptors, the alpha-receptor, and it has an anti-oxidant effect (it blocks several free radical generating enzyme systems that are pathologically upregulated in CHF). Coreg blocks the alpha-receptor, dilating peripheral arteries, but because it also blocks the beta-receptor, there is no secondary rise in HR. The only biochemical down-side of Coreg is that it blocks the beta-2 receptor as well as the beta-1, but this down-side is outweighed by its other benefits. Coreg is the Cadillac of beta-blocker therapy in CHF, but it also costs 10 times more than Metoprolol. Studies have shown that Coreg is superior, and worth the extra cost, in patients with advanced CHF who are not responding well to Metoprolol. In these patients, ejection fraction (the percentage of blood ejected by the heart with each beat, a measure of pump function) will rise and filling pressures (blood volume and blood pressure within the heart) will fall, following a switch from Metoprolol to Coreg. When beginning treatment on a new patient, I will recommend Coreg if it is covered by your insurer and if your co-pay is reasonable. If not, then I will use Metoprolol, saving you $100/month to be better spent on treatments that insurers should but do not cover, such as magnesium, thiamine, ribose, and Co-enzyme Q10. If you are struggling on Metoprolol, and have the financial means, then I will encourage you to switch from Metoprolol to Coreg. The side-effects of these agents include fatigue, an excessive reduction in HR and BP if I overdose you, depression, erectile dysfunction, and insomnia. We thus start with low doses and slowly work them up. The starting dose of Coreg is 3.1 mg twice a day, advancing over several months, as tolerated by the patient, to a maximum of 25-50 mg twice a day. Metoprolol is begun at 12.5 mg twice a day, advancing slowly to 50 mg twice daily. When applying the principles of beta-blockade, we must remember that the studies demonstrating the effectiveness of beta-blockade in CHF were all carried out in symptomatic patients who had first been stabilized with diuretic therapy, an ACE-Inhibitor, and often Digoxin. Beta-blockade is never a first line or stand alone treatment of CHF. The value of beta-blockade in asymptomatic patients with pump dysfunction is not clear.
Angiotensin Converting Enzyme Inhibition (ACEI) - Angiotensin I, elaborated by the kidney in response to renin, is a weak vasoconstrictor. Angiotensin I can be converted by Angiotensin Converting Enzyme (ACE) into Angiotensin II, a potent vasoconstrictor and free radical generating inflammatory mediator. You want a lot of Angiotensin II if you are combating reduced forward flow on the basis of blood loss or sepsis, but in the condition of chronic CHF Angiotensin II does a lot of damage. Angiotensin Converting Enzyme Inhibitors (ACEIs) block the generation of Angiotensin II, blocking the production of this most damaging cardiovascular whip. The first generation of ACEIs blocked ACE only within the circulation; these agents remain useful in hypertension, and do help in CHF. The newer, lipophilic (fat soluble), Tissue Specific ACEIs block ACE in the circulation and within arterial cells. Intracellular Angiotensin II degrades Nitric Oxide (NO) and generates damaging free radicals. The fat soluble ACEIs also block the degradation of Bradykinin, an angiochemical that upregulates NO and beneficial prostaglandin (the beneficial angiochemicals made from fish oil) production. These fat soluble, or Tissue Specific ACEIs improve endothelial function, the most important determinant of outcome in the patient with CHF or coronary disease. These agents (Quinapril, trade name Accupril, and Ramipril, trade name Altace) provide all the standard benefits of ACEI, and much more. It makes little sense not to use a Tissue Specific ACEI in patients with CHF (they also have an advantage in hypertension and coronary disease). Now that Quinapril is available in generic form, there is no reason to stick with the older agents. The side-effects of ACEI consist of fatigue if we over shoot and lower your BP too much, kidney dysfunction if you become dehydrated or if pre-existent kidney disease is present, and hyperkalemia, our term for an elevated serum potassium level. Diuretic agents waste potassium, such that potassium supplementation is typically required. If we add a ACEI, which tends to retain potassium, we will likely have to back off on your potassium supplement. A non-threatening but bothersome side-effect of ACEI therapy is a dry cough. This is due to Bradykinin, which is upregulated during ACEI therapy. Bradykinin is a beneficial angiochemical, but the cough can be a real irritation. If this occurs we will switch you from ACEI to an ARB (angiotensin receptor blocking agent). ARBs don't block ACE or the formation of Angiotensin II, but by blocking the Angiotensin II receptor on cardiovascular cells, they block many but not all of the deleterious effects of Angiotensin II. ARBs do not upregulate Bradykinin, and they do not significantly improve endothelial function. I use ARBs only in patients who cannot tolerate ACEIs. One rare but important side-effect of ACEI is angioedema, swelling of the tongue and throat that can compromise upper airway function. Angioedema is rare, but if this occurs you should never again take one of these agents. The starting dose of Quinapril is 10-20 mg/day, increasing as required to a maximum of 80 mg. Cost will be in the $30-50/month. Ramipril is begun at 2.5-5 mg/day, with a maximal dose of 20 mg/day. Ramipril is still on patent and consequently cost will be greater.
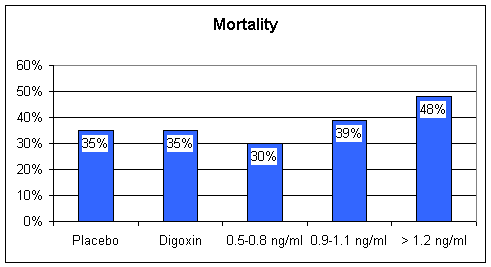
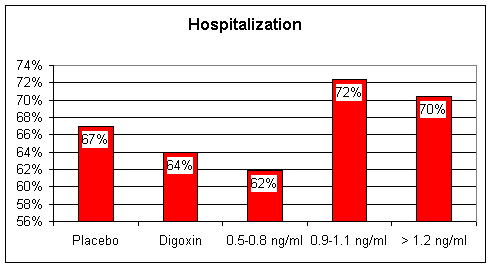
Digoxin - William Withering in the 1700s found that tea brewed from the Foxglove plant was of therapeutic value in the treatment of heart failure, then termed "dropsy". Withering also observed that too much Foxglove tea killed the patient. The active chemical in Foxglove is digitalis, referred to as Digoxin (trade name Lanoxin). Digoxin increases the "contractility" of the heart muscle; with digoxin on board, the weak, failing heart beats stronger. In patients with atrial fibrillation, a rapid, uncoordinated heart rhythm where the atrial storage chambers no longer beat in rhythm with the ventricular pumping chambers, digoxin will predictably slow the heart rate, decrease the heart's requirement for oxygenated blood, and improve cardiac performance. Digoxin also down-regulates the baroreceptor reflex, and by this mechanism contributes to neuroendocrine blockade. Baroreceptors within the arterial wall sense the forward flow of arterial blood. In heart failure, forward flow is compromised, the baroreceptors are stimulated, and the sympathetic nervous system releases adrenalin and nor-adrenalin. Digoxin blunts this response, sparing the heart from this chemical whipping. Digoxin has a narrow "therapeutic window". Just as Withering observed, a little digoxin is of value to the patient with CHF, but too much is lethal. 15 years ago, when Digoxin and Lasix were the only CHF treatments available, we would often "push Digoxin" too far. Patients would come in with high Digoxin levels, and toxic cardiac rhythms. Emergency pacemaker placement might be required for slow rates, or electrical cardioversion for rapid arrhythmia - I spent many a night dealing with digital toxicity. Dehydration, potassium and magnesium depletion, all side-effects of diuretic therapy, predispose to Digitalis toxicity, and all of our CHF patients were on diuretics at high dose. Now that the newer neuroendocrine blocking agents are available, many physicians have stopped using Digoxin in the treatment of CHF. While all Cardiologists agree that Digoxin is useful to provide heart rate control in the patient with CHF and atrial fibrillation, opinion is divided as to whether Digoxin is useful in CHF alone. A large, multi-center study Digoxin vs. placebo in CHF patients otherwise treated with diuretics and ACEI demonstrated that Digoxin administered at low dose, achieving a blood level of 0.5-0.8 ng/ml (the prior "therapeutic range was > 1.2ng/ml) helped keep patients out of the hospital and had a beneficial effect on mortality. Higher dose (more accurately stated higher blood level) Digoxin therapy decreased hospitalization rate but had now effect on mortality. I will recommend low dose Digoxin to patients with CHF and pump dysfunction if I do not think they are at high risk for Digitalis toxicity, and I will aim for a level between 0.5 and 0.8, but this is not a first line maneuver. If I think that you are at risk for digitalis toxicity (unstable potassium level or fluctuating kidney chemistries) then I will recommend that you not take this agent. The typical dose of Digoxin in CHF is 0.125 mg/day. The cost here is approximately $8/month.
Aldosterone Blockade - Angiotensin II , the potent, free radical generating vasoconstrictor, also stimulates the adrenal gland to secrete Aldosterone. Aldosterone stimulates the kidney to retain sodium and water, while wasting potassium and magnesium. Aldosterone is also manufactured in the adrenal gland, vasculature, and within the heart by an Angiotensin II independent mechanism, so blocking Angiotensin II generation with an ACEI (Angiotensin Converting Enzyme Inhibitor) alone will not fully block Aldosterone up-regulation and its deleterious effect on the patient with heart failure. Aldosterone worsens endothelial function, a disaster for the patient with CHF, particularly if co-existent coronary disease is present. Aldosterone affects the autonomic nervous system, stimulating the adrenergic fight or flight response and lowering heart rate variability. In the heart Aldosterone renders the muscle fibers stiff, culminating in myocardial fibrosis; susceptibility to arrhythmia is also increased. Aldosterone is good to have around if you are bleeding to death or septic following an encounter with another saber toothed tiger, but in CHF it does you no good. Spironolactone blocks the action of Aldosterone. Spironolactone blocks the Aldosterone receptor and blocks the Aldosterone effect. Spironolactone rights all the wrongs created by Aldosterone. Spironolactone improves endothelial function, the symptomatic status of CHF patients, and lowers mortality. The starting dose is 25 mg/day, advancing to 50 mg if necessary. Spironolactone by itself is a relatively weak diuretic, but it will potentiate the action of other diuretic agents, so we will watch your BP and kidney chemistries when we add Spironolactone in to your CHF regimen. Spironolactone retains potassium, a plus in the patient on diuretic therapy, but the combination of Spironolactone and an ACEI can sometimes raise the potassium level too high, so we need to watch for this as well. One nuisance side-effect of Spironolactone is breast tissue stimulation in men, leading to breast enlargement, termed gynecomastia (Digoxin can rarely do this as well). Eplerenone (trade name Inspra), will not cause gynecomastia, but otherwise has all the beneficial actions of Spironolactone. A study of Inspra in patients with recent heart attack and heart failure demonstrated a symptomatic and mortality benefit. Inspra at the standard dose of 50 mg/day costs $125/month. Spironolactone at 25 mg/day costs $18/month; thus we start with Spironolactone.
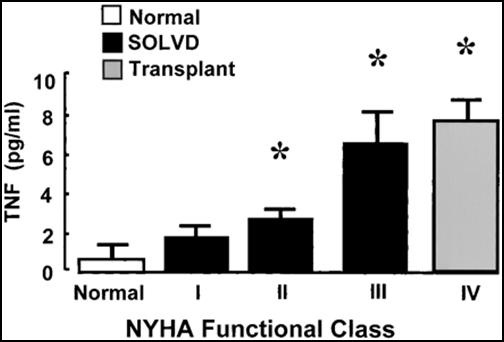
Pentoxifylline for TNF Blockade - CHF is an inflammatory condition. Patients will be red in the face; their pulse rate is often elevated and sometimes they will run a temperature. Patients with advanced heart failure will appear wasted, as if they had a tumor. We have observed that patients in active CHF appear as if they are infected, but what could infection have to do with CHF, a disease of the heart and circulation? We do know that infection can damage the heart; bacteremia (bacteria in the blood stream) lowers the ejection fraction. It's not the infection itself that compromises cardiac function, rather it is the immune system's inflammatory response to infection, any infection, that damages the heart. All of the inflammatory mediators that rise in infection (Interleukin-6 and C-Reactive Protein, to name a few) can damage the heart and the vasculature. Tumor Necrosis Factor-Alpha, referred to as TNF, is the most important offender. The presence of bacteria or bacterial cell membrane components within the circulation leads to the elaboration of TNF, which in turns stimulates an inflammatory immune response to kill the invading organisms. TNF also damages the heart, causing the myocytes to falter and then to commit apoptosis (premature cell suicide). During a brief infection, TNF levels are elevated only transiently, and the heart comes through unscathed, while chronic infections are more likely to compromise the heart. Now, consider the issue of fluid retention in the patient with CHF. Fluid retained behind the right heart manifests itself most obviously as ankle edema. What you don't see is congestion in the liver, and in the veins draining the GI tract. Bowel wall edema can impair absorption of the drugs we give you, and it allows bacteria to translocate from the lumen of the bowel, where they cause no harm, into the vasculature, where they illicit an inflammatory response and upregulate TNF production. As CHF is a chronic condition, TNF is chronically upregulated, and heart cells die off. As more and more heart cells die, cardiac pumping function worsens, the degree of CHF and bowel wall edema increases, more bacteria leak in, and down goes the patient. Multiple studies have demonstrated an inverse relationship between TNF levels and functional status and outcome in patients with CHF.
So what can we do about this? Of interest, TNF also plays a role in
Rheumatoid Arthritis, and TNF blocking therapy with Etenercept (trade name
Enbrel) is effective in reducing Rheumatoid symptoms. The side-effects of
Enbrel, which binds to TNF and basically lowers the level of free TNF to zero,
are an increased susceptibility to infections, and an increased cardiovascular
event rate (Why should this be - if TNF kills heart cells shouldn't TNF lowering
therapy be beneficial?). TNF blockade with Enbrel, in theory, should be of
value to the patient with CHF, but several studies of Enbrel in the treatment of
CHF showed either no benefit or a worse outcome compared with placebo therapy.
It turns out that the healthy heart needs a little TNF. At low levels TNF
plays a healthy, homeostatic role. It's only at high, especially
chronically high levels that TNF damages the heart. Enbrel wipes out TNF
completely, taking away its beneficial as well as its deleterious effects.
Pentoxifylline (trade name Trental) was introduced 15 years ago in the treatment
of claudication, effort related leg muscle pain due to lower extremity vascular
insufficiency. It is now off patent and thus not pushed by the drug
companies. It turns out that Pentoxifylline modulates the production
(technically it modulates the transcription of the gene coding for TNF - MME
probably does the same thing and we are working on demonstrating this) of TNF.
If TNF production is pathologically up-regulated by chronic infection,
Pentoxifylline will lower TNF, but if TNF production is normal, Pentoxifylline
will not lower TNF levels to the floor, as will Enbrel. In theory,
Pentoxifylline should help in CHF without an Enbrel-like down side.
Randomized, placebo controlled, multicenter studies of Pentoxifylline in CHF due
to coronary insufficiency/prior heart attack, dilated cardiomyopathy, peri-partum cardiomyopathy, and CHF due to chronic hypertension have been
carried out, and all showed a major improvement in ejection fraction, functional
status, treadmill  time, quality of life, and mortality. Pentoxifylline at
the usual dose of 400 mg three times a day costs around $18/month.
However, if you read review articles in the standard cardiology journals, you
will read that CHF therapies directed at TNF are uniformly ineffective.
They will not mention the positive studies on Pentoxifylline, even if they were
published in the same journal earlier. If your other physicians see that I
put you on Pentoxifylline and you do not have peripheral vascular disease, they
will question why. The disregard of Pentoxifylline in medical journals and
within the discipline of Cardiology is a bizarre oddity. Could it be that
the journals derive so much of their income from drug company advertising that
they don't want to showcase reduced CHF mortality for $18/month? Maybe the
experts who author the review articles are receiving too much grant money from
the drug industry. I really don't understand this, but I am quite liberal
with the prescription pad when it comes to writing for Pentoxifylline in my
patients with CHF.
time, quality of life, and mortality. Pentoxifylline at
the usual dose of 400 mg three times a day costs around $18/month.
However, if you read review articles in the standard cardiology journals, you
will read that CHF therapies directed at TNF are uniformly ineffective.
They will not mention the positive studies on Pentoxifylline, even if they were
published in the same journal earlier. If your other physicians see that I
put you on Pentoxifylline and you do not have peripheral vascular disease, they
will question why. The disregard of Pentoxifylline in medical journals and
within the discipline of Cardiology is a bizarre oddity. Could it be that
the journals derive so much of their income from drug company advertising that
they don't want to showcase reduced CHF mortality for $18/month? Maybe the
experts who author the review articles are receiving too much grant money from
the drug industry. I really don't understand this, but I am quite liberal
with the prescription pad when it comes to writing for Pentoxifylline in my
patients with CHF.
Statins for CHF - Statin drugs lower cholesterol, but that is not how they help in atherosclerotic vascular disease, and in CHF they help you despite their cholesterol lowering effect. Actually, there is an inverse relationship between serum cholesterol and mortality in patients with heart failure. Stated otherwise, the lower your cholesterol, the more likely you are to die. Look at the graphic below:
Here
it looks like cholesterol is somehow protective against mortality in CHF.
There are several minor and one major 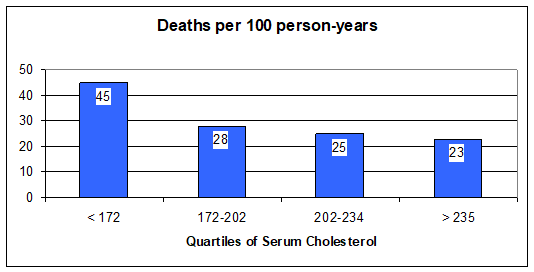 reason for this:
reason for this:
A. Cholesterol in an antioxidant. When a free radical oxidizes
cholesterol, a free radical is quenched; it is used up. As free radical
mediated inflammation plays a deleterious role in CHF, having cholesterol on
hand to serve as a free radical neutralizer is beneficial.
B. Cholesterol transports fat soluble Co-Enzyme Q10, and several other
cardioprotective antioxidants within the circulation; the patient with CHF needs
this transport mechanism.
C. Cholesterol probably plays a role in the transport of heavy metals out
of the body; as heavy metals play a role in CHF, this transport system may be
important.
D. Statins don't work by lowering cholesterol. I'm not saying that the reduction in serum cholesterol brought out by statin therapy does not play some useful role, but the major benefit of statin therapy is an anti-inflammatory one. Statins blocks HMG-CoA Reductase, an enzyme that plays a role in the synthesis of cholesterol. HMG-CoA Reductase doesn't block the final step, the synthesis of cholesterol, but rather a step that creates phosphorylated isoprenoids, farnesyl-PP and geranygeranyl-PP. While these compounds with interesting names do serve as intermediaries in the manufacture of cholesterol, they are more important in cell signaling. Farnesyl-PP and geranygeranyl-PP can transfer their isoprenoid groups to other signaling proteins, such as Rac, Rho, Ras, Rab, and laminin. When these proteins are prenylated, they turn on a host of inflammatory and free-radical generating enzyme systems. These in turn promote coronary atherosclerosis and its complications, and in the patient with heart failure, they promote apoptosis and an enhanced inflammatory response (you now know more about statin therapy than most doctors do - they call this type of knowledge "Alternative Medicine").
So cholesterol is protective, but its intermediates, intermediates blunted by statin therapy, are deleterious. Statin therapy also lowers Co-Enzyme Q10 levels by 50% (Co-Enzyme Q10 is manufactured along the same path as is cholesterol). Co-Enzyme Q10 deficiency can cause CHF, and supplementation is therapeutic. On one hand, in theory, statin therapy should help in CHF, while on the other it should hurt. What happens in reality? Let's find out. In one study patients with symptomatic CHF and an average ejection fraction of 33% (normal is 50-55%), nearly all of whom were on b-blockers, ACEIs, Digoxin, and a diuretic, were randomized to receive Simvastatin (trade name Zocor) 10 mg/day or placebo in double-blind fashion. The Zocor group did better. TNF, Interleukin-6, and B-Natriuretic Peptide levels fell while endothelial function improved. Functional class and ejection fraction improved.
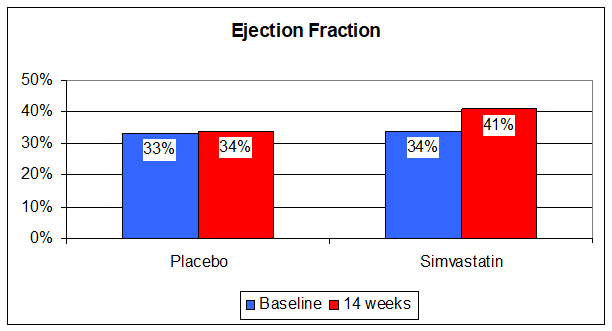

So the beneficial, anti-inflammatory effects of low dose statin therapy outweighed the deleterious effects (cholesterol reduction and Co-Enzyme Q10 depletion). It is thus my practice to treat CHF patients, particularly if their inflammatory markers are elevated, with low dose statin therapy, but only with concomitant Co-Enzyme Q10 supplementation. High dose statin therapy in patients with CHF is biologically insane. Even low dose statin therapy causes impaired cardiac diastolic dysfunction (impaired relaxation, poor cardiac filling) in people with normal hearts, diastolic dysfunction that responds to Co-Enzyme Q10 supplementation. I frequently see middle aged cardiomyopathy patients with advanced pump dysfunction (an ejection fraction in the 20s) and totally normal coronary arteries, who are on high dose statin therapy because their cholesterol is elevated. Now, if you are 60 years old and you have a high cholesterol, but your arteries are totally normal, you don't need to be a rocket scientist (by the way, I have a rocket scientist in my practice, so I get to use this joke all the time) to understand that the high cholesterol is not causing any trouble. We doctors need to realize that we are treating cholesterol not because it is high, but to prevent the high cholesterol from layering out as arterial plaque. If your arteries are normal at age 60 they are not going to plug up any time soon, and probably never, but if your ejection fraction is only 20%, if something intelligent is not done soon you will soon be dead. I explain the physiology to these patients, reduce their statin dose to 10 mg., and load them up with Co-Enzyme Q10 (along with the usual nutritional supplements and the neuroendocrine blockade drugs discussed above), and they get better. Their ejection fractions rise, and their other doctors become angry - they really do - I'm not kidding here.
Summary - Drug therapy for CHF emphasizes Neuroendocrine Blockade. Diuretic therapy is used to resolve symptoms due to fluid overload, to lower intra-cardiac filling pressures such that the heart is not mechanically stressed, and to prevent bowel wall edema, which might compromise the absorption of orally administered drugs and nutritional agents, and to prevent bacterial translocation across the bowel wall into the circulation. ACEI with a Tissue Specific agent is used in a nearly universal fashion; ARBS are used only when the patient cannot tolerate ACEI due to cough. In the absence of kidney dysfunction or other factors that might predispose to digitalis toxicity, low dose digoxin makes sense. b-blockade, utilizing either Coreg or Metoprolol is phased in to the patients regimen, as is Spironolactone. Pentoxifylline is important, especially if the patient's TNF level is elevated, along with low dose statin therapy, always accompanied by Co-Enzyme Q10 supplementation. Nutritional approaches to CHF are covered elsewhere on this site, as are mechanical measures, including EECP, MME, and dual; chamber pacemaker implantation. All play a role in the Integrative approach to Cardiology, the approach that we offer at Comprehensive Heart Care.
James C. Roberts MD FACC
1/01/07