
Return to Medical Topics Home Page
Return to CHC Home Page
Atherosclerotic Oxidative Stress
A Maladaptive Immune System Response to Perceived Intimal Infection
James C. Roberts MD FACC FAARFM
Oxidative Stress occurs when superoxide generation exceeds the ability of our innate enzymatic and diet-derived antioxidant defenses to neutralize this reactive oxygen species (ROS) and its physiologic second messenger H202, allowing the generation of longer-lived and vasculopathic ROS and RNS (reactive nitrogen species). Oxidative stress is the driving force of atherosclerosis, a pathophysiology that we can understand and contain.
This section covers the causes and consequences of intimal oxidative stress, as well as strategies to prevent and counter its adverse effects (particularly the Th1/Th17 led acquired immune system attack against oxidized LDL and other altered intimal proteins). Let’s start with a step by step review of atherosclerosis, with particular focus on steps mediated by oxidative and (secondary) inflammatory stress and immune dysregulation.
Focal Endothelial Activation and Lipid Infiltration
Atherosclerosis begins when apo-B 100 containing lipoproteins (LDL, Lp(a), and remnants) infiltrate the artery wall at sites of endothelial activation. This focal perturbation in endothelial barrier function occurs beyond branch points, where low laminar flow combined with high oscillatory shear leads to reduced eNOS (generates nitric oxide) and Nrf-2 (antioxidant enzyme transcription) expression, combined with up regulated NADPH Oxidase, Xanthine Oxidase, and Ang II activity. An intimal local environment is thus created where reactive oxygen and nitrogen derived species (ROS and RNS) are not counterbalanced by nitric oxide and intrinsic enzymatic antioxidant defenses. As flotsam and jetsam accumulate beyond a bend in the river, so will atherosclerosis initiate beyond arterial branch points.
Lipid particles traverse the endothelial cell monolayer via diffusion. The greater the number of lipid particles, and the smaller their particle size, the greater will be the level of passive lipid translocation. Systemic hypertension favors this process, as will other factors, including toxins, that promote systemic endothelial activation (adaptive in our response to true infection but when maladaptively present in atherosclerosis we term this phenomenon endothelial dysfunction).
This initial lipid intimal translocation is not by itself immuno-pathologic, as apo-B 100 is composed of amino acid sequences to which the innate and acquired immune system is not intolerant. LDL itself is food. It is a transport vehicle for the biosynthetic raw material cholesterol. Every cell of the intima expresses the molecular LDL receptor, at a level commensurate with its perceived need for free cholesterol. If the cell perceives a need for free cholesterol, the receptor will be expressed. If not, the LDL receptor is not expressed. LDL will not be taken up (your stomach is full so you put down your knife and fork).
Once oxidized, LDL cannot be taken up via the molecular LDL receptor; it is no longer biochemically useful. Thus, LDL carries with it antioxidant protection (lipid soluble antioxidants such as tocopherol, beta-carotene, and Co-Enzyme Q10, which can be "recharged" by water soluble Vitamin C). This defense is typically sufficient to protect LDL within the relatively reductive plasma environment. The extracellular intimal compartment, however, is more pro-oxidative (a thousand times more so than plasma). Standard LDL antioxidant defense levels may not provide sufficient protection. This should not be an issue, as "unneeded" LDL particles, those not ligated by an open LDL receptor, will freely diffuse back in to the circulation, and find an open LDL receptor elsewhere. In the absence of infection/inflammation, LDL will be internalized by cells only when and if it is needed for biosynthetic activity (steroid hormones, bile salts, Vitamin D synthesis, etc.).
Physiologic intimal LDL give and take, a demand and supply phenomena, has worked well for Man over our four-million-year history. Mother Nature promotes hyperlipidemia and focal infiltration of lipids beyond homeostatic synthetic need only when these processes are needed to defend against Man’s natural predator, which is infection. Primitive Man experienced oxidative and inflammatory stress only in relation to infection. Modern Man experiences a pseudo-infectious pathophysiology in a progressive, age-related fashion, in relation to our ROS-generating diet, life style, expanding waist line, and cumulative toxin burden – thus lipid metabolism is deranged!
To fight infection, we need more cholesterol (leukocyte cell membrane synthesis), we need ROS and RNS "bullets" (fired by wbcs to kill the invaders), and we need an activated endothelium (expressing adhesion molecules and elaborating chemotactic signals to pull immune cells in to the breach). HMG Co-A Reductase, the rate-limiting enzyme in cholesterol biosynthesis, thus up regulates, generating copious quantities of needed cholesterol, along with isoprenoid signaling molecules, which upregulate NADPH Oxidase, our most powerful superoxide generator, and down regulate eNOS, activating the endothelium. Oxidative stress means that "we are at war"!
Oxidative and inflammatory byproducts of Western living, false flags for chronic infection, thus lead to an age-related increase in circulating cholesterol, a progressively activated and thus leaky endothelium, and a biochemical milieu characterized by oxidative stress and dysregulated (Th1/Th17 rich and Treg poor) immune activation.
Thus, we have hyperlipidemia and endothelial activation, particularly beyond branch points. Lipids will infiltrate the endothelium at these stress points, but again, if they are not needed, if there are no open LDL receptors, then they will diffuse out. With respect to pathological atherosclerosis, the first thing to "really go wrong" is lipid retention within the subendothelial space. Retained lipid particles cannot diffuse out, and are thus subject to pre-atherosclerotic oxidative modification.
LDL Modification, Trapping, and Oxidation
LDL cholesterol is composed of cholesterol and cholesterol ester (food), containing a small number of single use non-enzymatic antioxidants (protectors), surrounded by the apo-B 100 protein, which is studded with phosphatidylcholine molecules. The rheological characteristics of a given LDL particle relate to the status of its surface phospholipids (the initial composition of which, in turn, relates to relative dietary intake of saturated vs. unsaturated fatty acids).
Altered (termed minimally oxidized, or MM-LDL) and now immobilized LDL particles will be subject to further oxidative alteration by free radical species generated within the sub-intimal space (initially with superoxide generated during normal oxidative metabolism and later with superoxide and secondary ROS/RNS species generated by pathologically up regulated NADPH Oxidase (NOX), Xanthine Oxidase (XO), Angiotensin II – Angiotensin Receptor type 1 (AT1R) trafficking, and HMG Co-A Reductase expression.
Double bonds on LDL particle surface polyunsaturated fatty acids (PUFAs), susceptible to oxidative alteration, convert into reactive aldehydes, which form adducts with exposed lysine and arginine of the apolipoprotein B100, altering LDL particle configuration and charge. The now heightened negative charge of the oxidized LDL particle (oxLDL) allows tighter binding to the positive charge of proteoglycan sulfate and carboxyl groups as well as to the scavenger receptor of tissue mononuclear cells, favoring its immobilization and uptake, respectively. Of greater consequence, this "acquired mutation" of apo B100 creates a protein sequence, or more precisely a protein shape, to which developing T lymphocytes were not exposed to during their maturation within the thymus. Oxidized apo B100 thus appears as a foreign molecule, an invader, a microbial "look alike" that must be killed or neutralized by the full force of Mother Nature’s anti-infectious defense mechanisms.
Oxidized LDL Phagocytosis and Innate Immune System Activation
At this point we have trapped LDL molecules that are being oxidized, and an activated, nitric acid poor, endothelial surface that is non-specifically pulling in monocytes (termed macrophages upon entrance into the intima). Macrophages, as do all cells of the intima, bear LDL receptors. Oxidized LDL is not ligated by the native LDL receptor; it no longer fits. Rather the scavenger receptor expressed by mononuclear cells recognizes oxidized LDL as a microbe or cellular debris. Oxidized LDL is thus taken in. Having captured this non-native and thus threatening particle, the macrophage activates. We kill microbes with superoxide and downstream ROS and RNS, and thus their production within the macrophage is increased. More scavenger receptors are elaborated. The activated macrophage will also express non-phagocytic threat receptors (such as TLR4, the Toll-Like Receptor, and cytokine receptors), such that it can better sense its local environment and carry out its search and destroy mission. ROS that cross into the local environment serve to hasten oxidation of adjacent immobilized but not-yet-oxidized LDL particles. The activated macrophage releases chemotactic molecules (such as MCP-1, monocyte chemotactic protein), creating a chemoattractant trail to lead mononuclear cells that ligated endothelial adhesion molecules to actively translocate to the point of perceived infection, and activation signals, such as MCSF (monocyte colony stimulating factor), stimulating the translocating mononuclear cells to "lock and load", up regulating ROS/RNS generation and scavenger and threat receptor expression.
Intimal infection has been diagnosed. The endothelium further activates. Monocytes, along with second wave monocular defenders (dendritic cells and T memory cells) swarm in, more cholesterol is oxidized, and more oxidized cholesterol is phagocytosed. At this point, the activated macrophages contain more cholesterol than it needs for biosynthetic purposes, and thus its native LDL receptor should be withdrawn. That is not the case. As will be discussed in more detail later, inflammatory cytokines, the products of Nuclear Factor Kappa Beta (NF-k B) translocation, stimulate the conversion of free cholesterol into cholesterol ester (storing fuel for the infectious winter to come). As expression of the native LDL receptor relates to the level of free, not esterified cholesterol within the cell, the LDL receptor remains expressed even though the activated macrophage is chock full of phagocytosed oxidized LDL. The activated macrophage, eager to non-specifically rid the local environment of all potential threats, not just the threat it perceives from oxidized LDL, begins to pinocytose adjacent molecules, including non-oxidized LDL particles.
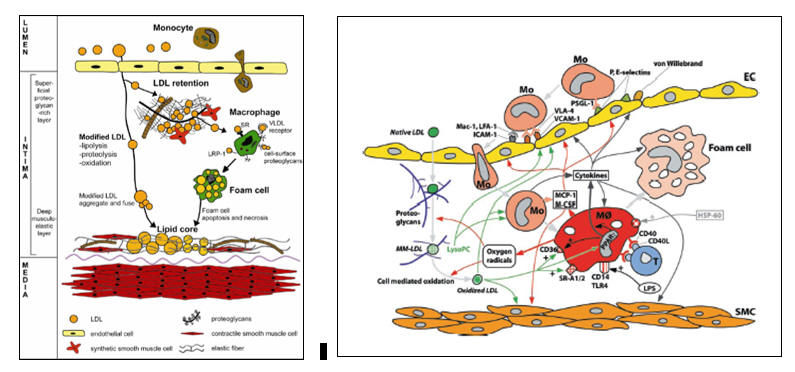
We now have an activated macrophage, loaded to the gills with oxidized cholesterol, sending out distress signals to attract and activate fellow first line defenders. While later arriving, innate immune defenders can leave this inflamed intimal environment to warn the adaptive immune system as to the specific nature of the oxLDL threat, the early arriving, and now cholesterol choked macrophage inactivates, transforming into a lipid laded foam cell. Apoptosis and coalescences of foam cells creates the fatty streak, the initial histologic manifestation of atherosclerosis.
This process, of course, is reversible. When a microbial threat has been neutralized, a wave of non-activated macrophages (M2 macrophages) will infiltrate the previously inflamed region and phagocytose the dead first line defenders, allowing function and histology to return to normal (termed catabasis). This process is programmed to occur. When Mother Nature initiates an inflammatory response, the biological clock begins to tick, and a few days later the immune response shifts from search and destroy to inflammation resolution, as the cytokine milieu shifts from Th1/Th17 (Il-1b , TNFa , Il-6, Il-17, Il-23), to Treg (Il-10 and TGF-b ). This programmed shift from infiltration to catabasis will occur, of course, only if the infectious threat is indeed neutralized. If microbes continue to breach the endothelial barrier, the inflammatory response is not called off, and inflammation resolution will not occur. In the setting of human atherosclerosis, if the level of lipid particles diffusing in to vulnerable endothelial sites decrease, then the foam cells will be resorbed. In contrast, if cholesterol infiltration continues (the situation in Industrialized Man), then catabasis will not occur, more foam cells will be created, apoptosis will give way to tissue necrosis, and we now have a mass of coalesced lipid droplets and crystals within the vascular wall, the lipid core of the developing plaque.
Even before we get to this point, of atheroma development, and well afterwards, we do have a means of removing cholesterol, and reversing LDL oxidation, within the intimal environment. Mother Nature (evolution or the creator, depending on your perspective) knew that our corrective response to endothelial microbial breach in the setting of infection-induced hyperlipidemia could lead to macrophage lipid engorgement and subsequent apoptosis. Thus she (or he, again depending on your perspective) created the HDL system. The HDL-associated enzyme Lecithin Cholesterol Acyl-Transferase (LCAT) esterifies free or de-esterified cholesterol from mononuclear and intimal cells with linoleic acid, and then loads up the newly formed cholesterol-linoleate into the HDL particle, for transport back to the liver. Paraoxonase, another HDL-associated enzyme, removes lipid peroxides from mononuclear cells, intimal cells, and lipoproteins, essentially reversing LDL oxidation (and why stimulating HDL reverse cholesterol transport and anti-oxidant function protects against disease progression and adverse events).
Anatomic atherosclerosis begins with the fatty streak. If cholesterol levels fall, then catabasis and reverse cholesterol transport can occur, and the fatty streak will resorb. If cholesterol continues to breach the endothelium, then more mononuclear cells will be brought in, and the fatty streak gives way to the slowly growing atheroma. The focal absence of endothelial derived nitric oxide allows vascular smooth muscle cells to proliferate and migrate in to the endothelial zone, adding bulk to the atheroma. At some point the lesion will become visible on angiography.
So far, we have confined ourselves to the innate immune response, how pattern receptors on mononuclear cells react to perceived environmental threat. But atherosclerosis is a maladaptive response of the acquired immune system to perceived infection of the arterial wall with oxidized LDL and other perceived non-native entities. How do we go from LDL oxidation to the Th1/Th17 rich, Treg poor intimal environment where Interferon-gamma stimulates macrophages to release matrix metalloproteinases to degrade the fibrous cap and precipitate an acute coronary event? A review of immune system dynamics is thus warranted.
Immune System Basics
Immune system effector cells, both innate and adaptive, originate within the bone marrow from a common hematopoietic progenitor stem cell line. Innate immune cells sense threat in a non-specific fashion; they respond to abnormal shapes. They sound an alarm, and stimulate an adaptive, or acquired immune response against a specific invader (in true infection, a snippet of bacterial protein; in atherosclerosis, initially an 8-20 amino acid in length snippet of oxidized apoB 100).
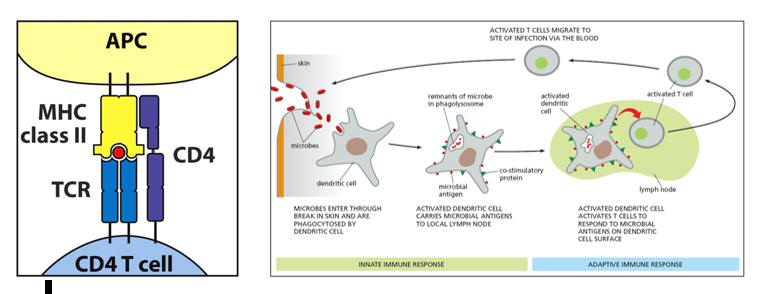
Innate immune cells sample the environment. A large surface area facilitates this function, and thus they send out cytoplasmic pseudopods (hence the generic term dendritic cells), coated with receptors that recognize non-native shapes. PAMPs (Pathogen Activated Molecular Receptors) recognize lipopolysaccharide of gram-negative microbes and like molecules expressed by gram positive bacteria. DAMPs (Damage Activated Molecular Receptors) recognize debris of apoptotic cells or particulate matter and oxLDL. In fact, oxLDL is the most common DAMP. Monocytes serve as circulating dendritic cells. Upon entry into the intima or other tissue compartments they transform into macrophages. Kupfer cells (liver), microglia (brain) Langerhans's cells (skin) are all dendritic cells that have specialized to sense threats found within these specific tissue environments.
So, a microbe breaches our skin, or an oxidized LDL protein forms within the intima. PAMPs on the first in dendritic cell ligates the non-native shape in question. The structure is then phagocytosed into a lysosome, where it is degraded into its component parts, including snippets of its protein structure. Having captured an invader, the roving dendritic cell now migrates to the nearest lymph node or lymph organ (liver or spleen), to inform the adaptive immune system as to the specific threat faced, aiming to initiate a rapidly amplifying immune response (antibodies and cytokine elaborating T helper cells) directed against the specific invader. In this process the dendritic cell, functionally an antigen capture cell, converts itself into an antigen presenting cell (APC), prepared to present what it caught on a MHC II molecule, to display co-stimulatory molecules, and to secrete co-stimulatory cytokines, all in an effort to awaken a naïve T helper cell, to serve its one and only function, to activate, proliferate, and then direct an overwhelming immune response to a specific threat.
The lymphocyte population of the acquired immune response consists of B cells and T cells. B cells generate antibodies, only in response to T cell instruction, aiming to immobilize or damage invaders that are not destroyed via the mechanism of macrophage phagocytosis. B cells play little role in atherosclerosis and will not be discussed further. Nascent T cells leave the bone marrow, and migrate to the thymus, where they are "educated". Within the thymus, T cells will be exposed to antigenic determinants (8-20 amino acid in length snippets of native protein) that they will see within their lives within the specific human. T cells that react with normal proteins will be culled via apoptosis. The trillion different T cells that survive thymic maturation can "read" antigenic determinants presented on MHC molecules. No surviving T cells should react to a self-molecule (clinical auto-immunity occurs only when a self-molecule has been altered, such as in myocardial infarction, or when chronic, unrelenting oxidative and inflammatory stress over stimulates the immune system, such that it begins to indiscriminately misrecognize self as foreign). Following thymic maturation, naïve, non-activated T cells migrate from the thymus to peripheral lymph organs, where they do nothing until they are awakened from biochemical slumber by an activated innate immune cell that bears an antigenic determinant, on a MHC II molecule, that is a specific match for the T cell receptor of the resting T cell.
Innate immune cells thus awaken dormant T helper cells by presenting antigenic determinants on an MHC II molecule. What are MHC molecules? What are T cell receptors? How does the acquired immune system activate to legitimate infection, or pathologically to oxidized LDL or troponin?
MHC (Major Histo Compatibility) molecules are flag poles that can fly different flags. Cell membrane bound MHC molecules "present" 12-20 amino acid in length protein snippets, designed to inform adjacent or roving T cells as to the proteins contained within them.
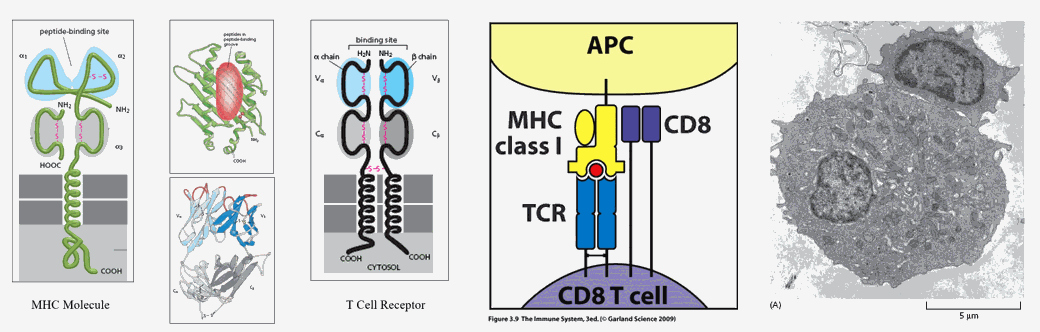
Non-immune, somatic cells bear MHC I molecules. Their job is to inform and activate natural killer T cells and CD8 self-surveillance T cells if their cell has undergone malignant transformation or viral invasion. Our cells generate specific proteins, in relation to which genes are being transcribed within the nucleus, when specific mRNA molecules are translated into specific amino acid sequences at the level of the ribosome. Transcribed proteins contribute to cell structure and function. After a period of time each cell protein will be degraded, thus allowing for an adaptive turn over in cellular protein, in relation to which genes are being translated, related to differing intracellular conditions, related to differing signals the cell is receiving from its outside local and systemic environment. Intracellular proteins are degraded within proteosomes, which will cut the protein into 12-20 amino acid in length antigenic determinants, which are then attached to MHC I molecules to be expressed on the cell membrane. CD8 and T killer cells are monitoring this situation. If all intracellular proteins are native, then the antigenic determinants presented on MHC I molecules are something that they have seen before, something to which they are tolerant, and no action is taken.
However, if the cell has been hijacked, say by a virus, and is now cranking out non-native, viral proteins, or if the cell has undergone malignant conversion and is generating inappropriate proteins, then non-native antigenic determinants will be expressed on the MHC I molecules. Roving natural killer and CD8 T cells will recognize this anomaly, bind to the now rogue cell within an immune synapse, and then destroy it (clinical cancer occurs only when this monitoring process breaks down).
Antigen Capture and Presentation
The initial immune synapse, at least one hundred "lock in key" MHC II – antigenic determinant – T cell receptor molecular couplings between the antigen presenting cell (APC) and naïve T cell, is necessary to initiate an acquired immune response directed against the specific antigenic determinant (and the microbe from which it was derived), but by itself is not sufficient. The APC must elaborate co-stimulator molecules and co-stimulatory cytokines to "fully wake up" the resting naive T cell. Stated otherwise, the APC must be "alarmed and angry" if it to fully "lock and load" the resting T cell (in this fashion, we will not mount an acquired immune response to a normal protein that was accidentally internalized by a first line defender). Interleukins released by the APC awakens the slumbering naïve T cell. As it is activating, the previously naïve T cell will likewise release cytokines to further activate the APC to stimulate its own clonal proliferation. The greater the level of mutual co-stimulation, the greater will be the level of T cell activation and clonal proliferation.
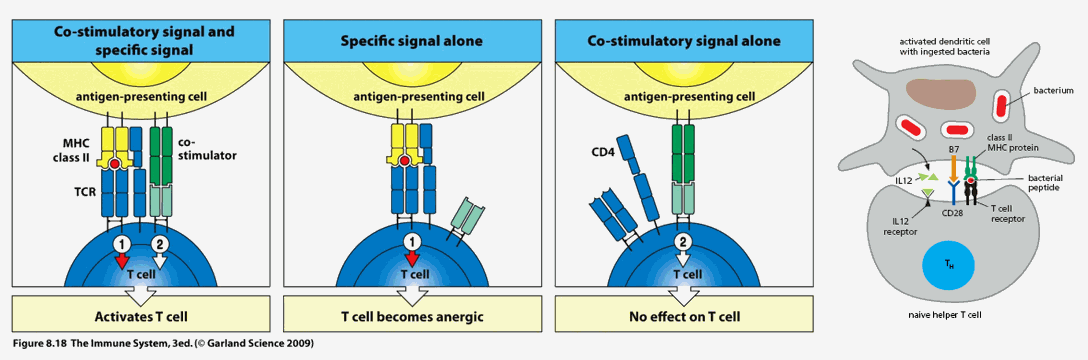
CD80 and CD86 (often referred to as B7) on the APC will interact with CD28 on the resting T cell. The now activating T cell will express CD40L, which will interact with CD40 on the APC. CD4 on the T cell (T helper cells are all CD4 while T cells that eliminate rogue native cells are termed CD8) coordinates the interaction between and antigenic determinant bearing MHC II molecule on the APC and the TCR (T cell receptor) on the resting T cell.
Mother Nature thus created a number of checks and balances (which we can understand and therapeutically manipulate) with respect to whether or not the immune system will activate against a specific antigenic determinant, as well as to the direction and magnitude of any specific immune response to follow.
Naïve T cells activate in to one of four lineages. If an antigen capture cell is bearing news of a large invader, such as a parasite, that cannot be phagocytosed by immune cells, then it will elaborate cytokine Il-4, which directs the activating T cell to differentiate within the Th2 lineage. Th2 cells instruct specific clones of B cells to generate antibodies to neutralize and kill the critter. If the first line defender instead has captured a bacterium which can be phagocytosed by mononuclear cells, then it will activate within the Th1 or Th17 framework. These T helper cells secrete stimulatory cytokines and bear membrane signaling molecules designed to activate mononuclear cells to phagocytose and kill bacterial invaders. Not all antigens captured by innate immune cells are an appropriate target of an immune response. Antigens derived from inhaled or ingested molecules ideally should not lead to an immune response; in this situation we would become immunological reactive to molecules that we breath in or take in within our diet. Thus, the biochemical milieu within immune organs in the upper respiratory and GI tract promote activation of T cells into the Treg lineage. Treg cell membrane expressed CTLA-4 binds tightly to co-stimulatory molecules expressed by the APC, turning down the APC’s activation state. Soluble Treg generated cytokines such as Il-10 and TGFb turn down APC and T helper cell activity (stable plaques contain Treg cells; this inhibition is lost in unstable lesions).
Above and beyond the characteristic of the protein snippet presented to the resting, inactive T cell, and the location at which the immune synapse is occurring, the internal biochemical milieu of the lymph organ plays a role in determining the route into which the T cell will develop. Vitamin D, for example, alters the characteristics of the antigen presenting cell, such that the co-stimulating molecules that it elaborates will encourage the newly stimulated T helper cell to take an immune down regulating Treg course (consider the link between Vitamin D sufficiency/insufficiency and the incidence of multiple sclerosis, which involves auto-immune attack against self-protein).
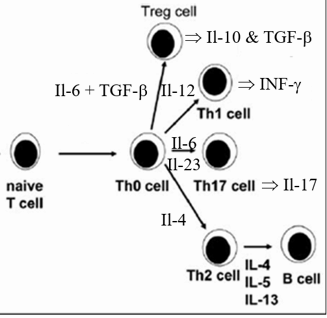
The cytokine and redox status of the lymph organ will greatly influence the
lineage of a newly activated T cell. One’s immune history, in a sense,
determines one’s immune future. If you are a chronic allergy sufferer, you bear
Th2 cells that secrete Il-4 and stimulate B cells to make antibodies
(unfortunately for you) to the pollen that you just inhaled (not enough Treg
cells and too many Th2 cells and thus allergy occurs). Allergy begets more
allergy, as the more Il-4 within the lymph organs, the more likely will you
mount an undesirable Th2 response to newly presented protein snippets.
Conversely, lots of Il-12 within the lymph organ skews new immune responses to
newly presented antigenic determinants down the Th1 pathway. If you suffer from
recurrent bacterial invasion, as did primitive man, then the lymph node is rich
in Il-12, and you tend toward a Th1 response. Recurrent infection adds Il-17 to
the mix, stimulating T cells to mature within the Th17 lineage (essentially a
more powerful version of the TH1 cell).
Oxidative stress trumps all other T lineage determinants, and promotes
maturation down the Th1 and Th17 pathways. The immune response to oxidized LDL,
and other abnormal intimal proteins, as well as to troponin and other myocardial
molecules to which a deleterious T cell response occurs in heart failure, is Th1
and Th17 driven. CV disease is characterized by skewing of the immune response,
toward Th1/Th17 and away from Th2/Treg. Oxidative stress initially skews the
immune response, these T cell release inflammatory mediators that lead to more
oxidative stress, and the viscous, self-stimulating cycle of oxidative stress
and immune dysregualtion that drives CV disease follows.
Once activated by an upregulated innate immune cell that bears an antigenic determinant that matches its T cell receptor, the T cell assumes its role as a T effector cell. The cell line proliferates (generating Il-2 and a high affinity Il-2 receptor, leading to oligoclonal proliferation of this specific T helper cell line) and the daughter cells migrate to the periphery, aiming to kill their target (microbe in infection and oxidized LDL in atherosclerosis) at the site of initial stimulation of the innate immune system and at all other points in the body.
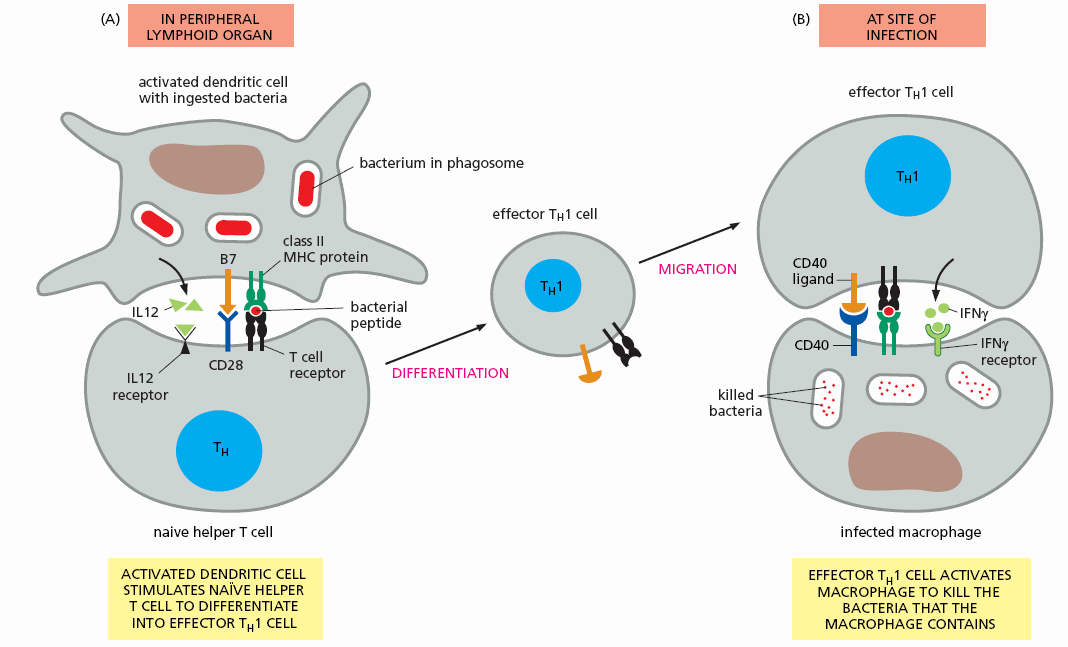
The previously naive T cell bearing a T cell receptor specific for a protein snippet of a microbe (in this example a bacteria or a snippet of oxLDL) has activated and proliferated. Millions if not billions of these activated helper T helper cells are soon swarming the circulation, seeking to help macrophages and other innate defenders phagocytose and kill the invaders (before they can kill us).
T Helper Cell Homing and Reactivation
How do the T cells know where to go? While the initially activated (within the lymph organ closest to the site of initial infection) Th1 helper cell can access any site within the vasculature, it makes more sense if they can be drawn in to the region where the innate immune system initially identified the focal breach. This will be an area where activated immune cells (macrophages and dendritic cells) have generated ROS/RNS and inflammatory cytokines, such that the local endothelium has activated, generating chemokines (MCP-1) to pull circulating mononuclear cells towards the site of infection as well as adhesion molecules (ICAM and VCAM) to tether the mononuclear cells to the site of endothelial activation, making it easier for the cells to enter the subintimal space (where their target, invading bacteria or oxLDL snippets, reside). The first T helper cell bearing a T cell receptor specific to a protein snippet derived from the invader (microbe or oxLDL) has now been drawn in to the area of infection.
Along with the newly activated T helper cell line, our circulation also contains T memory cells, allowing the acquired immune system to rapidly respond to recurrent infection. After a microbe has been eradicated (a joint effort between the innate and acquired immune systems), local ROS/RNS generation tails off, T cells specific to the target cease to proliferate, and most then die of senescence. A few persist, as T memory cells. They "remember" the threat, continue to bear TCRs specific to the threat (the specific antigenic determinant), and exist in a dormant state. However, upon reinfection, re-exposure to "their" remembered antigenic determinant, they can fire up rapidly, undergo oligoclonal proliferation and "return to the breach". With your initial exposure to a specific microbe you may be sick for a week, the time needed to mount an effective acquired immune response to the specific organism. Upon re-exposure, you may be sick for one-two days or not at all, as you now possess an at least partial "immunity" to the invader.
Vaccination creates T memory cells to specific potential invaders. Antigenic determinates derived from microbes or inactivated microbes are administered along with adjuvants (pro-inflammatory substances such a thimerasol) that stimulate innate immune cells to activate, internalize the exogenous protein, and then transform in to an APC and initiate an acquired immune response to the potential invader. If the CDC accurately predicts which flu strains will dominate in a given year, then they will create a flu vaccine that generates T memory cells that will recognize and eradicate the incoming viruses. If they predict incorrectly, the flu vaccine will not protect you, as the flu virus is constantly mutating, trying to outwit Mankind’s defenses (our T memory cell repertoire).
The circulation contains T memory cells, related to prior invasion, and newly activated T helper cells specific for the new infection (or pseudo-infection with oxLDL in atherosclerosis or altered myocardial molecules in heart failure). All of these T cells, along with circulating dendritic cells, will be attracted to and pulled into a region of endothelial activation, where infection resides, or (pathologically) at coronary branch points where ROS/RNS generation has activated the endothelium, allowing entry of LDL molecules that subsequently became trapped, oxidized, and internalized by mononuclear cells. All of these infiltrating T cells will sample the local intimal environment. If there are no activated intimal cells (macrophages or dendritic cells initially but within the oxidative milieu of an activated plaque smooth muscle and endothelial cells can transform in to antigen presenting cells) expressing "their" specific antigenic determinant, then they will return to the circulation, to see if they can be useful elsewhere.
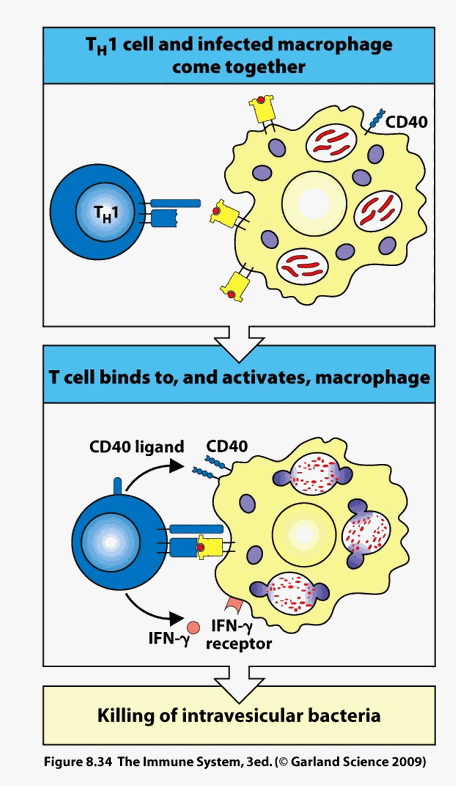
Thus, the number and type of immuno-reactive antigenic determinates within a local intimal environment (and the corresponding number and type of ligating T cells) will determine its overall activation state. If the molecules within the intima are normal, self in nature, then its T cell repertoire will consist of Treg cells, which release Il-10 and TGFB, which down regulate, or temper the level of immune activation. An activated plaque contains few Treg cells, and 20-40 clones of T helper cells that have undergone local oligomeric proliferation in response to reactivation against 20-40 specific antigenic determinants. The local milieu will be inflammatory (Th1 and Th17 cytokines) and oxidative (these cytokines stimulate endothelial, smooth muscle cells, and infiltrating mononuclear cells to generate ROS/RNS).
The Links Between Infection and Atherosclerosis
20-40 clones of T helper cells reacting to 20-40 specific antigenic determinants! What are the determinants, from what proteins were they derived, and how did they get into the intima? We can qualitatively identify the T cell repertoire in plasma, within a plaque, within any region of the body. The T cells are harvested, and in vitro reacted with antigenic determinants derived from specific microbes or specific proteins. If T cells proliferation occurs (you measure radiolabeled thymidine incorporation) you can infer that T cells bearing TCRs specific for that microbe/protein snippet were present in the tissue sampled (and if you are dealing with an atherosclerotic plaque, that the same microbe/protein snippet was present within the plaque, at some time point). In the absence of infection, plasma will contain small numbers of T memory cells specific to any infection that you have experienced, a bell-shaped curve or polyclonal distribution of lymphocytes. If you contract an infection, say pneumonia, you will see skewing of the T cell repertoire, with oligoclonal proliferation of T memory cells specific to the invader or previously naïve, newly activated T helper cells reacting to a newly identified microbial antigenic determinant. As the infection clears, T cells specific to the microbe will die of senescence, and the polyclonal distribution will return. The T cell repertoire within a plaque relates to the age of the lesion, infectious activity elsewhere in the body, and local and systemic levels of ROS/RNS and cytokine milieu. 10% of the T cells within any plaque will react to oxLDL. A similar number will react to intimal structural proteins such as beta-2 glycoprotein and heat shock protein (hsp). Stressed cells, human and microbial, translocate mitochondrial hsps to the cell membrane, aiming to stabilize their cytoskeleton (you are under attack, so circle your wagons). Intimal hsp expression is negligible within the healthy intima, low level within an early lesion, and extensive within a complicated lesion. But these are self-proteins. Why should they be subject to an acquired immune response? These proteins can be oxidized, rendered immunogenic, just as in the case of oxidized LDL. Also, in the presence of overwhelming ROS/RNS/cytokine stimulation, the frenzied acquired immune system can "break tolerance", misrecognizing self-proteins (that are at the wrong place at the wrong time) as non-self (consider the link between intestinal hyperpermeability and auto-immune disease; with chronic cytokine stimulation the immune system starts making mistakes).
If you carry out incidental atherectomy of a non-inflamed, non-culprit, 60% RCA narrowing in your patient who presents with chest pain and ST segment depression on the basis of an inflamed 95% LAD lesion, you will find that the T cell repertoire, and immune histology of these two lesions are quite different. To paraphrase Tip O’Neil, atherosclerotic immune activation is local. The smooth non-culprit narrowing will contain macrophages, dendritic cells, and T helper cells, certainly some specific for oxLDL protein and other altered intimal proteins. Within a stable plaque, there will also be Treg cells, elaborating Il-10 and TGBF, aiming to neutralize the interferon-gamma and other Th1 cytokines being released from plaque Th1 helper cells, keeping plaque inflammation in check. Nitric oxide inhibits vascular smooth muscle cell (VSMC) growth and proliferation. Nitric oxide was lost long age, and proliferated VSMCs, along with fibroblasts, have generated a stabilizing fibrous cap, sequestering lesional atheromatous gruel and inflammatory activity from the vessel lumen. Such lesions, if they obstruct the lumen sufficiently, may produce effort-induced ischemia. If adjacent vessels are not severely diseased, and if local nitric oxide is available, then the pressure differential between the patent and diseased vessel will lead to the elaboration of a protective collateral network. Stable plaques, characterized by a low ROS/RNS/inflammatory cytokine burden, will not rupture/erode to precipitate ischemic injury (as a therapeutic corollary, if we can convert an inflamed plaque in to a stable plaque, ischemic event risk will attenuate).
The culprit plaque demonstrates a quite different immune histology. Along with T helper cells specific for oxLDL and other altered intimal proteins, you will find 20-40 clones of T cells specific to a wide variety of microbes. You are aware of the link between infectious history and atherosclerosis. The greater the number of microbes to which you display immune experience (reactive T cells or IgG antibody levels), the greater is your atherosclerotic risk (and risk of disease recurrence following revascularization).
How does this link work? Are the microbes invading the intima, or is the link indirect? Recall that an active plaque is constantly elaborating chemotactic signals and adhesion molecules, aiming to pull in mononuclear help (Mother Nature is fighting what she perceives as infection, initially with oxLDL). Let’s say you bear a chronic bacterial infection (gum disease being the most common). Mononuclear cells infiltrate the gum tissue, gobble up the corresponding microbes, and digest them within phagolysosomes. Some hightail it to the nearest lymph node, generating a proliferation of Th1/Th17 cells specific to the invader, while some return to the circulation. If that mononuclear cell happens to traverse a coronary vessel containing an active plaque it may be non-specifically pulled in to the lesion. There it may display gum microbe antigenic determinants to gum microbe sensitized T cells (T memory cells) that have also been non-specifically pulled in to the lesion, reactivating them to battle mode. As long as gum disease is present and the plaque remains active, more and more gum bacterial antigenic material will enter the plaque, and thus the immune system will be mounting an inflammatory response against gum disease within the active plaque. Nearly all plaques contain T cells sensitized to gingival invaders such as P. gingivalis. Common pulmonary pathogens such as C. pneumonia and M. pneumonia, frequently encountered viruses such as EBV and CMV, and GI and GU microbes will also be represented. In fact, virtually any infection can do this, such as bacterial, viral, fungal, TB, parasite and other infections. As long as the plaque is active, focal infection elsewhere will contribute to plaque immune dysregualtion. Keep this principle in mind the next time you attend a meeting in Las Vegas, where you might be tempted to interact with the wrong sort of people without proper protection. Do you want those sorts of microbial proteins within your coronary intima? The link between infection and atherosclerosis led to antibiotic intervention trials, which for the most part were unsuccessful. This is because the bacteria within the intima are long dead. Within the coronary vasculature T cells are reactivating to their antigenic determinants, not to the microbes themselves. Preventing chronic infection (and thus chronic immune stimulation) by killing the microbes where they reside, however, will be helpful (thus resolution of gum disease reduces your coronary risk). Also, recurrent bacterial infection will lead to skewing of the immune response towards Th1/Th17 and away from Th2/Treg, creating a cytokine milieu that drives atherosclerosis and heart failure. Heat shock protein (HSP) molecular mimicry is another link between extra-vascular infection and atherosclerosis. The HSP concept has worked well in evolution, for man and microbes. The HSP amino acid sequence of man and his common pathogens are little different. In response to infection, let’s say with C. pneumonia, we will mount an immune response against antigenic determinants derived for this organism. The invaders feel the heat, and thus elaborate hsps on their outer surface. We then respond by generating an immune response against C. pneumonia hsp, hastening clearance of the invader. Healthy endothelial cells do not express hsp, unhealthy cells (smokers, diabetics, individuals bearing a toxin burden) do, as do atherosclerotic cells, at a level commensurate with the degree of local immune dysregulation. Smokers experience more MIs in the winter than in the summer, in part related to immune cross-reactivity between endothelial and microbial hsps.
Plaque Activation
The atheromatous core of a stable plaque is contained by a fibrous cap. In response to dietary/life-style change and/or appropriate pharmaceutical/nutraceutical intervention, ROS/RNS generation will have curtailed, Treg cells will be neutralizing Th1/Th17 activity, and endothelial nitric oxide production may return. This plaque will not progress and it will not activate.
The active, or vulnerable plaque demonstrates quite dissimilar cytology and histology. The plaque will be infiltrated, particularly at its shoulder region, with activated mononuclear cells, all elaborating pro-inflammatory cytokines, ROS, and RNS. Plaque derived chemokines are pulling in mononuclear cells bearing antigenic determinants from distant sites; foreign wars are now being conducted locally, and we see oligoclonal proliferation of 20-40 clones or Th1/Th17 cell lines within the plaque. Ongoing, intense inflammation leads to Treg cell regression (we don’t want Treg cells turning down our Th1/Th17 defenses when we are fighting chronic infection). The character of the T cell response also changes. Recall that initial cell activation and T memory cell reactivation requires "lock in key" co-stimulatory as well specific antigenic determinant – T cell receptor interaction. In response to chronic re-activation to its specific antigenic determinant, T helper cells mutate such that their co-stimulatory receptor, CD 28, drops off. These cells (CD4+CD28null) spontaneously express Il-12 receptors, and may activate in response to their antigenic determinate, or simply to Il-12. Thus, immune activation elsewhere, which might lead to Il-12 generation, can activate these cells within the unstable plaque (another link between winter infection and MI). CD4+CD28null cells are long-lived, spontaneously generate interferon-gamma, and are cytotoxic to endothelial cells. These cells may egress the activated plaque, may infiltrate distant intimal sites, and "splash" the vasculature with interferon-gamma. If you bear a 50% carotid plaque, it is far more likely to itself activate over the two years following an ACS (acute coronary event) than over a corresponding time period pre-ACS (you can think of this as metastatic plaque activation). The greater the percentage of plaque and circulating T cells that are CD4+CD28null in character, the greater is the likelihood of subsequent ACS (and conversely, the greater will be your gain with anti-atherosclerotic therapy).
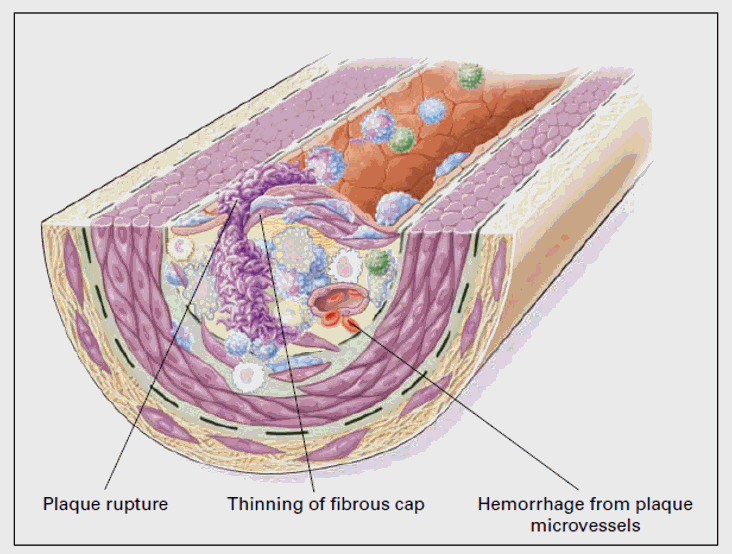
Atherosclerosis begins at points of intimal oxidative stress. Oxidative stress drives plaque progression and activation. Our goal, therapeutically, is not just to lower cholesterol (which rises with age in relation to oxidative status), but to lower oxidative stress, and with-it inflammation and the dysregulated, Th1/Th17 skewed immune response that drives vascular disease. A discussion of oxidative stress, how it relates to atherosclerotic risk factors, and how to attenuate it is thus in order.
The Oxidative Cascade
Generation of Superoxide (SO) and its second messenger Hydrogen Peroxide (H202,) are constitutive and necessary for normal cellular function. Their synthesis can be physiologically up regulated to deal with infection and trauma.
Oxidative stress (OS) occurs when the production of these reactive oxygen species (ROS) is chronically and inappropriately increased, overwhelming our innate enzyme based and dietary small molecule antioxidant defenses, allowing their conversion in to more long-lived and damaging ROS and reactive nitrogen species (RNS) such as peroxynitrite (ONOO), hydroxyl (OH), and hypochlorous acid (HOCL). When underdefended intimal ROS/RNS generation leads to endothelial activation, lipid trapping and oxidation, and secondary Th1/Th17 skewed immune dysregulation, we experience intimal oxidative distress, followed by atherosclerosis and its sequelae.
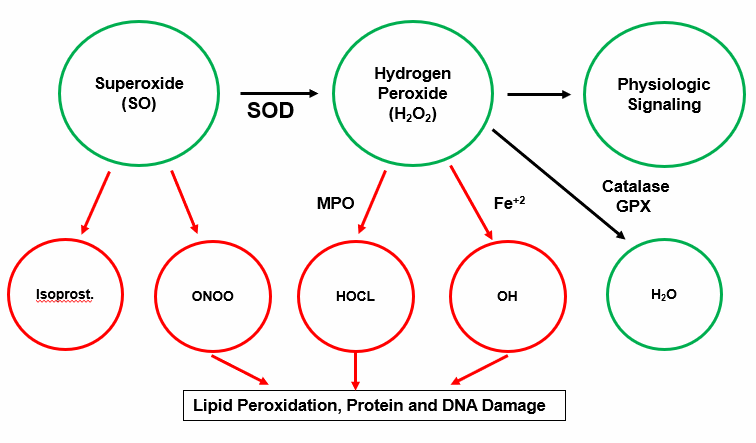
Free radical species, ROS and RNS, are reactive electrophiles; they are electronically imbalanced. ROS such as SO contain an unpaired electron within their outer orbital shell. Absent a neutralizing electron donor (an antioxidant), a ROS will snatch an electron from an adjacent structure, quenching its electron thirst, but creating a new radical species. A biochemical chain reaction occurs, with resultant damage to cellular lipids, proteins, and nucleic acids.
Antioxidant enzymes (mineral dependent) convert a ROS to a less toxic metabolite. "Spent" antioxidant enzymes must then be recharged by a secondary antioxidant enzyme, or be resynthesized. Diet derived antioxidant molecules "donate" an electron to the ROS, quenching its electron thirst, rendering it non-reactive. "Spent" antioxidants can be recharged by other antioxidants. Mother Nature designed us to maintain a dynamic and adaptive balance between ROS generation and anti-oxidant neutralization.
Primitive Man did not experience inappropriate or chronic oxidative stress. When faced with infection, microbicidal ROS/RNS generation was transiently increased, allowing for threat eradication. Modern Man is experiencing chronic oxidative stress, rarely due to chronic infection, but endemically due to errors of modern living, such as diabesity, intestinal hyperpermeability, chronic emotional stress, and age-related accumulation of radical producing metal and organic pollutant toxins, phenomena not anticipated by evolution. Risk factors for atherosclerosis are either a cause, or a consequence, of inappropriate ROS/RNS generation.
The primary source of constitutive SO production is the mitochondria, where 1-2% of inhaled oxygen is incompletely reduced to SO, designated as a free radical as SO contains an unpaired electron in it outer orbital shell (molecular oxygen contains two, and is chemically far less reactive). Housekeeping enzyme systems (protein generation, digestion, and nutrient assimilation) generate lesser quantities of SO. SO is short-lived, but if not converted into H202 by superoxide dismutase (SOD) it can react with nitric oxide (NO) to generate peroxynitrite (ONOO).
Intimal ONOO generation, outside of our response to infection, is 100% maladaptive, as are the other "atherosclerotic bastard" descendants of SO and H202 (OH, HOCL, and lipid radicals). They fill no homeostatic need, they are not neutralized by endogenous antioxidant enzymes, they oxidize indiscriminately, and in this manner initiate and drive atherosclerosis, malignancy, and neurodegenerative disease states.
As stated above, ROS generation is constitutive, a necessary byproduct of oxidative metabolism. Low level H202 trafficking is also necessary for multiple cellular housekeeping functions, including protein folding, and appropriate cell growth and differentiation. Mother Nature generates and needs ROS, at a low level tonically, and at a high level transiently, when dealing with infection. Keeping SO and H202 under control, and preventing their conversion into ONOO, OH, and HOCL, is the responsibility of our mineral-based antioxidant defense enzymes, backed up, as needed, by diet-derived small molecule antioxidants.
Superoxide Dismutase (SOD), present as three cell compartment specific isomers, rapidly converts SO into H202. If SO generation is not pathologically (or appropriately in infection) up regulated, SOD will convert SO in to H202, and ONOO will not be generated. Copper and zinc-dependent SOD1 neutralizes SO within the cytoplasm. Manganese-dependent SOD2 degrades SO that is generated within the mitochondria. SOD3 (often referred to as extracellular SOD) is present on the endothelial surface or released into the intimal space; SOD3 protects these compartments against NO to ONOO conversion. Adequate mineral nutriture (not the rule in Western Man) is obviously important here. Some, but not all, diet-derived antioxidants, if at the right place at the right time, may assist in SO neutralization (Vitamin C, Co-Enzyme Q10, and melatonin but not Vitamin E).
Conversion of SO into H202 is thus a good thing, protecting against ONOO formation, and we do need low concentration H202 for homeostatic signaling. Excessive SO to H202 trafficking must also be dealt with, and here Mother Nature has provided a redundant defense.
Single purpose and single use Catalase (CAT) rapidly neutralizes H202 in to H2O. New CAT enzymes then must be transcribed via the Nrf-2 pathway. Dual purpose Glutathione Peroxidase (GPX) transfers an electron from reduced glutathione (GSH) to H202, also generating H2O (GPX can also neutralize lipid radicals). Now ‘spent" or oxidized glutathione (GSSG) is "recharged" in to GSH by Glutathione Reductase (GSR), with an electron derived from NADPH, itself generated within the pentose phosphate biochemical pathway. A similar enzyme system, Peroxiredoxin (PRX), also neutralizes H2O, and is "recharged with an electron transferred from NADPH by the Thioredoxin (TRX) system.
If our enzymatic and small molecule antioxidant defense systems can not contain SO and H202, then bad things happen. Arachidonic acid (split off from LDL phosphatidylcholine by PLA2) can be non-enzymatically converted in to pro-inflammatory isoprostanes molecules. The Fenton reaction, catalyzed by reduced iron (Fe2+) or copper (Cu2+), converts SO and H202 into hydroxyl anion (OH), a ROS with 1000-fold more destructive power (why we wish to avoid iron overload, why pre-menopausal status confers protection against atherosclerosis, and why low dose Vitamin C, which reduces Fe3+ to Fe2+, might work against us in the situation of iron overload). OH readily abstracts an electron from the double bond of cell membrane or organelle membrane polyunsaturated fatty acids, to create a lipid peroxide radical, which reacts with an adjacent double bond, setting up a membrane damaging chain reaction (generating oxLDL, malondialdehyde, hydroxyenol, and other lab markers, and mediators, of oxidative distress).
Lipophilic Vitamin E (Tocopherols and Tocotrienols), found within the cell or organelle membrane, will react with lipid peroxides, fortunately 1000-fold faster than the lipid radical can react with an adjacent double bond, breaking the chain reaction. Spent Vitamin E will then be recharged by hydrophilic Vitamin C within the circulation or cytoplasm, and spent Vitamin C can be recharged by diet derived bioflavonoids.
Myeloperoxidase (MPO) converts H202 into microbiocidal hypochlorous acid (HOCL). HOCL, essentially bleach, is great to have around during infection, but in the setting of inappropriate oxidative stress, is particularly damaging. MPO expression is up regulated, in turn, by up regulated H202 expression, because, as you appreciate, Mother Nature regards high level H202 expression as a sign of infection. If MPO remains chronically elevated due to chronic infections, it will cause HDL to become dysfunctional, increase CHD plaque rupture, open calcium channels in the arteries and increase blood pressure.
Paraoxonase (generated in the liver and associated with HDL within the circulation) excises oxidized regions within cell membranes, and within LDL and HDL particles. Paraoxonase thus mediates the antioxidant, or oxLDL neutralizing effect, of HDL. MPO is a physiologic antagonist of PON, degrading PON and oxidizing HDL, while PON degrades MPO and reverses lipoprotein oxidation. PON activity is uniquely up regulated by Pomegranate juice, in part mediating the vasoprotective properties of this diet-derived polyphenol antioxidant.
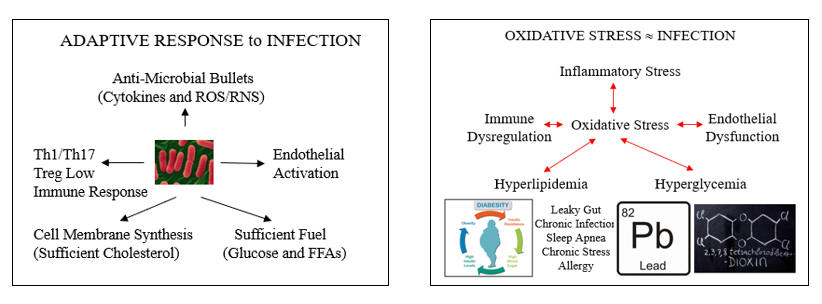
The cartoons below describe the appropriate systemic response to infection, and how oxidative and inflammatory cues, artifacts of Western living, have hijacked these systems to generate what we refer to as risk factors for atherosclerosis. Let’s explore these links, starting with endothelial dysfunction.
Oxidative Endothelial Dysfunction
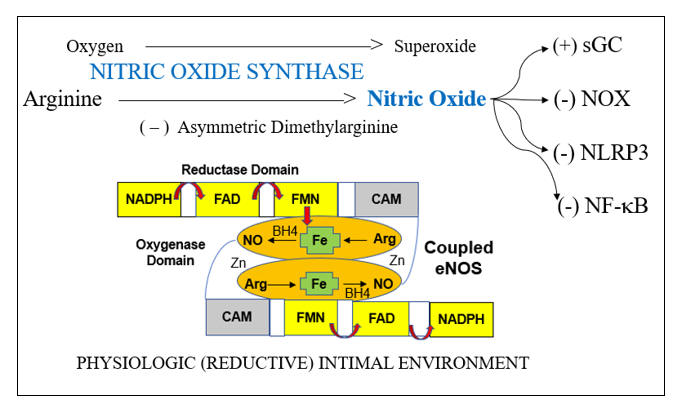
Endothelial dysfunction is a key determinant of outcome in all stages of atherosclerosis and in heart failure. This is because endothelial dysfunction is both a sign and a mediator of intimal and myocardial oxidative stress, the key driving force in CV disease. Before we discuss endothelial activation, an adaptive response to infection, let’s discuss how endothelial chemistry works in a physiologic, reductive environment (stated otherwise, when Mother Nature does not perceive infection). Endothelial Nitric Oxide Synthase (eNOS, or NOS3) converts arginine into Nitric Oxide (NO), which then diffuses within the cell and across the endothelial cell membrane, to activate soluble guanylate cyclase(sGC) in adjacent platelets and vascular smooth muscle cells. sGC in turn activates cAMP-dependent processes, which lead to vascular smooth muscle relaxation, with subsequent vasodilatation, and reduced platelet adherence and activation. NO inhibits vascular smooth muscle cell proliferation and hypertrophy (preventing hypertension and muscle cell encroachment within the intima). NO restrains the endothelial cell from elaborating adhesion molecules and chemoattactants signals that would pull leukocytes into the intima. NO also inhibits translocation of NF-kB, our primary inflammation amplification pathway, and generator of nascent Il-1b and Il-18, and activation of the NLRP3 Inflammasome, which activates the nascent cytokines and generates an inhibitor of endothelial tone. NADPH Oxidase (NOX), the key pathologic source of intimal superoxide (SO) is directly inhibited by NO. When eNOS is functioning properly we maintain appropriate vasodilation, platelets are not sticky, and we inhibit enzyme systems that would, if unrestrained, lead to intimal oxidative stress, inflammation, atherosclerosis, and plaque destabilization.

Within an oxidative intimal environment, not only are these vascular protections lost, but eNOS itself becomes a SO generator. Adaptively, in infection, and maladaptively, in the setting of chronic, acquired oxidative stress, SO generation is up regulated, outpacing the rate at which it can be converted to H2O2 by SOD. SO reacts with NO three times more rapidly than it does with SOD, creating the long-lived pro-oxidant peroxynitrite (ONOO). ONOO is not constitutively generated; it has no physiologic role other than to kill microbes. Thus, ONOO is not subject to degradation by our endogenous antioxidant enzymes systems. ONOO is not neutralized by Vit E, but it can be neutralized by taurine, N-acetyl cysteine, methyl-folate, and pharmaceutically, by hydralazine. ONOO, a reactive nitrogen species (RNS) will activate (via tyrosine nitration) pro-inflammatory enzyme systems. With respect to endothelial function, ONOO degrades BH4, the key co-factor of NOS. NOS then splits into its monomeric form, stops generating NO, and instead starts converting oxygen in to SO. Restraint on NOX is lost, leading to more SO, which along with the SO generated by now un coupled eNOS, combines with what little NO we have left to form more ONOO. Restraint of NF-k B (and a related inflammation amplification pathway, activator protein-1) is lost; thus, pro-inflammatory cytokines and endothelial adhesion molecules and chemokines will be elaborated. Restraint on the NLRP3 inflammasome is lost. This enzyme complex (discussed in the colchicine section) promotes maturation of nascent NF-k B generated Il-1b and Il-8, unleashing their inflammatory potential, and directly compromises endothelial tone. This intimal metabolic shift works in our favor when we are dealing with infection (the leading cause of death in Primitive Man), particularly if trauma is involved (Saber Tooth Tiger bite). Here you want vasoconstriction, platelet activation, endothelial activation within the infected region, and bullets (cytokines, SO, ONOO, HOCL, and OH). iNOS, or inducible NOS, is present in phagocytic cells and up regulates is response to infection, here generating NO to combine with SO to form ONOO, to help kill the invaders. Our clinical problem, in the treatment of CV disease, is that Mother Nature interprets oxidative stress as a sign of infection, and thus chronic, inappropriate oxidative stress converts eNOS into a mediator of inflammatory atherosclerosis.
eNOS activity is modulated by two other redox sensitive phenomena; differential shear stress and the ADMA to arginine ratio. Physiologic laminar shear stress (5-20 dynes/cm2) up regulates transcription of eNOS and Nrf-2 (which controls transcription of our innate antioxidant enzymes). Low laminar flow, just beyond branch points and on bends, creates focal points of reduced NO and antioxidant intimal protection. Oscillatory shear, present beyond branch points, up regulates expression of intimal SO generating enzymes systems (NADPH Oxidase and Xanthine Oxidase). Focal insufficiency of NO in the presence of excessive SO thus explains the focal initiation of atherosclerosis just beyond branch points, and secondarily along curves. Saphenous vein grafts last, on average, seven years, while 90% of Left Internal Mammary (LIMA) grafts remain patent at 10 years, also in relation to differential intimal redox status. The LIMA is a long straightaway, and thus non-hypertensive flow creates a reductive, atherosclerosis resistive LIMA intimal environment. The thin wall of the saphenous vein is designed for non-pulsatile venous flow; SO is not generated here and little NO is needed. Placing the SV within the high-pressure arterial circuit leads to up regulated SO generation, and thus rapidly developing SV graft atherosclerosis. While physiologic laminar flow promotes a reductive intimal environment, excessive laminar shear (> 20 dynes/cm2), on the basis of hypertension, turns up NADPH Oxidase, creating an oxidative intimal environment, essentially the link between hypertension and atherosclerosis.
eNOS enzymatic activity is also governed by the ratio between its raw material, the amino acid arginine, and its physiologic, competitive inhibitor, asymmetric dimethylarginine (ADMA). ADMA is generated at a constant, constitutive rate, in relation to protein degradation. ADMA is metabolized by Dimethylarginine Dimethylaminohydrolase (DDAH). When DDAH is functioning normally, ADMA is broken down rapidly, the ADMA to arginine ratio is low, and if eNOS has not been oxidatively inhibited, arginine will be converted in to NO. DDAH is inhibited by essentially all atherosclerotic risk factors, via the common mechanism of (you guessed it) oxidative stress. The more risk factors you bear, the more compromised will be DDAH function, the greater will be your ADMA to arginine ratio, the less NO you will generate. It is not the absolute level of ADMA that governs eNOS activity, but rather the ratio of ADMA to arginine; an imbalance here can be rectified with arginine supplementation.
Cumbersome methodologies have been utilized to measure endothelial function (intracoronary acetyl choline, forearm plethysmography, brachial artery flow-mediated vasodilation). Peripheral artery tonometry (EndoPAT) provides a low-cost (typically insurance-covered) office assessment of endothelial tone. Endothelial dysfunction is covered in greater detail elsewhere, but from the redox perspective we can address endothelial dysfunction with targeted antioxidant support (taurine and methyl-folate to neutralize ONOO and Vit C and N-acetyl cysteine to neutralize SO), neutralization of intimal SO generators (ARBs, HMG Co-A Reductase inhibitors, Berberine or Hydralazine to down regulate NADPH Oxidase, and Allopurinol to inhibit Xanthine Oxidase) and Arginine (2-4 grams tid). As we normalize endothelial function, we will also be turning down NF-k B, AP-1, NLRP3, and NADPH Oxidase activity.
Oxidative Stress Promotes NF-k B Translocation
Microbes divide rapidly, and thus we need to keep up, with the rapid generation of microbiocidal, pro-inflammatory cytokines and endothelial adhesion molecules. Our primary pathway of inflammation amplification is the Nuclear Factor Kappa Beta (NF-k B) transcription pathway. Under resting, reductive conditions, NF-k B is sequestered within the cytoplasm, complexed with IKBa (Inhibitor of Kappa Beta). Ligation of threat receptors on immune response cells activates a chain reaction of serine kinases (ERK 1/2, p38MAPK, c-Jun N-terminal kinase) which converge to activate Inhibitor of Kappa Beta Kinase (IKK) which degrades IKBa , allowing NF-k B to translocate to the nucleus and transcribe the appropriate defense molecules. LPS (bacterial cell wall lipopolysaccharide) appropriately activates this pathway, as will molecules found in the presence of legitimate infection (Il-1b , Ang II, CD40L).
These "threat molecules" (and other NF-k B agonists such as free fatty acids) up regulate pathologically, in relation to the pro-oxidant and pro-inflammatory cues present in Western Man (leaky gut, visceral adiposity, toxins, etc.) such that NF-k B is constantly on the move (NF-k B up regulates ten-fold within unstable plaques), cranking out pro-atherogenic molecules.
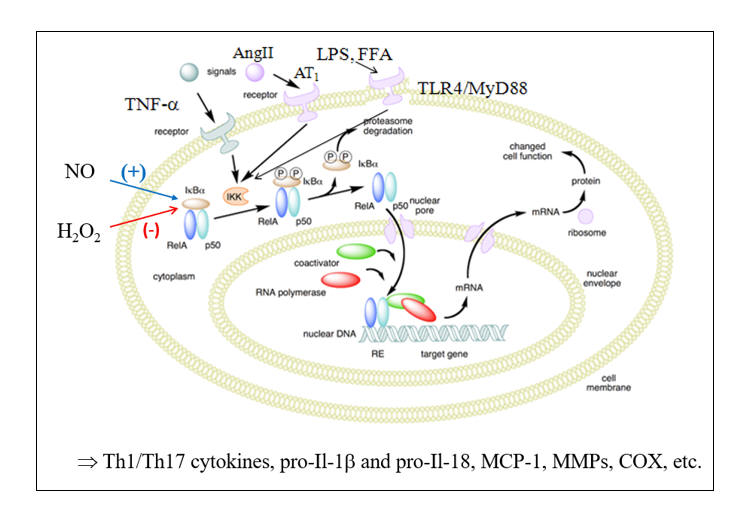
NO up regulates IKBa expression, and thus blunts NF-k B trafficking within the intima. This restraint is lost in the presence of endothelial dysfunction. H2O2, in excess, will activate IKK to degrade IKBa , shunting NF-k B to the nucleus. Stated otherwise, you do not need real infection, or the presence of infection signaling molecules, to activate the pro-inflammatory genes under NF-k B control; all you need is oxidative stress. Why? Primitive Man experienced oxidative stress only in relation to infection. Thus, our physiology interprets oxidative stress as "another infection" and translocates NF-k B, leading to the generation of 100s of pro-inflammatory mediators.
While episodic oxidative stress typically triggers a balancing, rebound generation of antioxidant molecules (via Nrf-2, to be discussed later), a downstream action of NF-k B is to inhibit Nrf-2 translocation (in legitimate infection we do not want antioxidant molecules; we want ROS/RNS bullets)
When circulating markers of inflammation, such as CRP or fibrinogen, are elevated, we often intervene with anti-inflammatory therapies, such as fish oil or turmeric. While these agents are helpful, a more expedient and complete approach would be to neutralize oxidative stress, as oxidative stress precedes and drives inflammatory stress.
The Hyperlipidemia of Pseudo-Infection
In response to legitimate infection, HMG-Co-A Reductase (the rate-limiting step in cholesterol generation) up regulates. This makes sense, as you need cholesterol to manufacture cell membranes for the rapidly proliferating leukocytic defenders. It also helps if you can activate the endothelium, making it easier for the defenders to access infected tissues, and to generate ROS/RNS bullets to kill the microbes.
HMG Co-A Reductase generates cholesterol through a system of intermediate molecules, including the isoprenoids farnesyl and geranylgeranyl pyrophosphate. They activate (via prenylation) the signaling molecules Rac, Rho, and Ras, which in turn upregulate SO production, activate the endothelium, and up regulate Ang II receptor expression. These actions generate more ROS, which in turn promotes NF-k B translocation, to generate more pro-inflammatory cytokines (a virtuous cycle to deal with infection).
This system worked great for primitive man. Genomic hyperlipidemia thus arose as a defense against perinatal sepsis, the leading cause of death in the history of mankind. A minority of us carry these traits, accounting for the infrequently encountered situation of familial hyperlipidemia (good for your ancestors but bad for you). As a corollary, if your bear sickle cell genes you will not die of malaria (Mother Nature never does anything without a good reason).
In the rest of us HMG Co-A Reductase is not genomically up regulated, and when we are young our cholesterol levels are low. However, in Western Man, cholesterol values start to rise in our 20s, and continue to rise with aging. Why is this occurring? What factors determine the level of expression of HMG Co-A Reductase?
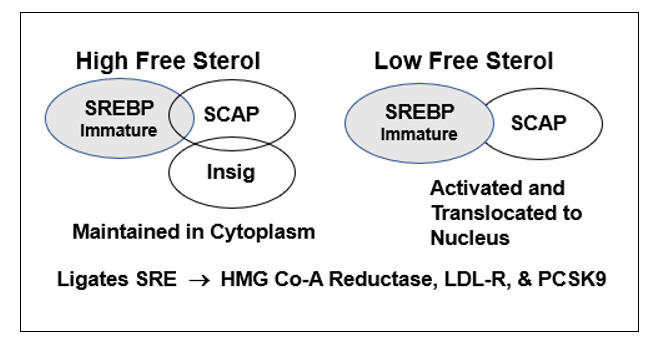
HMG-Co-A Reductase transcription is governed by the cell’s perceived need for free cholesterol. When intracellular free cholesterol is adequate, the free cholesterol sensor Insig sequesters SREBP (Sterol Regulating Element Binding Protein) within the Endoplasmic Reticulum. When free cholesterol is low, Insig releases SCAP (Sterol Regulating Element Binding Protein Cleavage Activating Protein) to transport SREBP to the Golgi apparatus, where it is enzymatically modified, promoting its translocation into the nucleus, where it binds the Sterol Regulator Element (SRE), the promoter site for the three key genomic sequences involved in cholesterol homeostasis.
Activation of the SRE leads to transcription of HMG Co-A Reductase, such that the cell can generate more cholesterol, and of LDL receptor protein, enabling the cell to pull more cholesterol out of the circulation. Cholesterol biosynthesis and uptake occurs mainly in the liver, and as Mother Nature doesn’t want the liver to hog all the cholesterol, SRE activation also leads to the generation of PCSK9 (Proprotein Convertase Subtilisin/Kexin type 9), a counterbalancing protein that degrades the hepatic LDL receptor, maintaining serum cholesterol to meet the needs of other cells.
In the presence of legitimate infection (lipopolysaccharide) or inflammation, Acetyl Co-A Acyl Transferase (ACAT) expression up regulates, promoting cholesterol esterification (to store cholesterol raw material for the upcoming infectious winter). As Insig senses only free cholesterol, HMG Co-A Reductase and PCSK9 transcription increase, even though total intracellular cholesterol is rising. Intracellular and circulating cholesterol thus rise with age. This doesn’t relate directly to dietary cholesterol (in our 20s we consumed pizza and beer and our cholesterol values were low) but rather to the progressive accumulation of pro-oxidant and pro-inflammatory phenomena (visceral fat, leaky gut, metals, organic pollutants, etc.) that lead to maladaptive HMG Co-A Reductase and PCSK9 transcription.
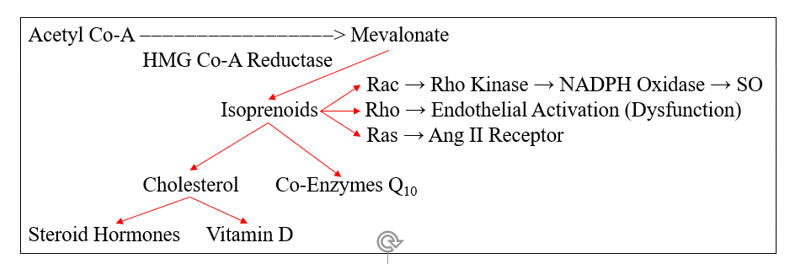
HMG Co-A Reductase does not generate cholesterol, it generates mevalonate, which converts to cholesterol, through the intermediate isoprenoid molecules farnesyl and geranylgeranyl pyrophosphate. These molecules activate (via prenylation) a series of GTPase signaling molecules that activate pathways critical to the response to infection, but which in our patients add to their oxidative/inflammatory burden.
Rac prenylation activates rho kinase, which translocates the NADPH Oxidase (NOX) regulatory element Rac to the cell membrane, enabling assembly and activation of NOX. Infection appropriate activation of phagocytic NOX is responsible for the SO "respiratory burst", while within the intima NOX generated SO is likely to react with NO to form ONOO, or within the diabese liver to downstream ROS/RNS that promote NF-k B translocation and heightened insulin insensitivity.
Rho prenylation down regulates eNOS activity, compromising NO generation, while Ras activation increases angiotensin receptor type I expression (ATR1), through which Ang II leads to NF-k B translocation and further NOX activation.
Avoiding or resolving the age and diet related oxidative/inflammatory cues that increase HMG Co-A Reductase transcription is the best approach here. But if the patient is unable or unwilling to take these steps, or if we do not have the luxury of time, then interventions to inhibit HMG Co-A Reductase activity will lower cholesterol and reduce trafficking through the NOX, ATR1, and NF-k B pathways. This can be achieved with statin drugs, nutraceutical "green statins" such as red yeast rice extract, bergamot, and amla, tocotrienols which hasten degradation of the enzyme, and AMP sensitive protein kinase (AMPK) agonists such as berberine, which physiologically (via phosphorylation) down regulate the enzyme. It really doesn’t matter how you inhibit flow through HMG Co-A Reductase, but if you do so cholesterol generation will fall, and oxidative stress and inflammation will attenuate.
The indication for intervention is not a specific cholesterol level, but rather the presence of inflammation and oxidative stress in the presence of atherosclerosis, endothelial dysfunction, or other factors suggesting that HMG Co-A Reductase is inappropriately up regulated. There will be little gain in 30-year-old women with an LDL of 130 and an otherwise pristine risk profile, but tremendous gain in her diabese grandfather who sustained his second MI due to graft failure, whose LDL is at a similar level. On the negative side, Co-Enzyme Q, steroid hormone, Vit D, and Vit K generation will also be compromised. Thus, if we intervene against HMG Co-A Reductase, we must also be ready to measure and replace these physiologic substances. We also need to remember that Mother Nature created countless other oxidative stress and inflammation up regulation pathways, so if we rely on statin therapy alone, we will not be covering all the necessary bases of atherosclerosis protection. As a consequence of HMG Co-A Reductase inhibition, PCSK transcription will inevitably increase (why you have to keep increasing the statin dose). Berberine blunts this process, and will thus synergize nicely with any HMG Co-A Reductase inhibition strategy (stated otherwise you can get the job done with a lower statin drug dose).
Oxidative Stress Promotes Insulin Insensitivity and Hyperglycemia
Insulin mediates and governs cellular glucose homeostasis. Insulin ligation of the Insulin Receptor leads to tyrosine phosphorylation of the Insulin Receptor Substrate (IRS) and downstream signaling molecules (PI3-K/Akt), leading to appropriate glucose uptake and utilization. Serine Kinases (such as IKK, the same molecule that shoots Nf-k B to the nucleus), up regulated in relation to oxidative and inflammatory stress, mediate antagonistic serine phosphorylation of IRS molecules, leading to insulin insensitivity, impaired glucose utilization, and hyperglycemia. This is helpful when fighting infection (leukocytes need glucose), but chronic disruption of insulin signaling leads to the progressive hyperglycemia, diabesity, and hyperlipidemia that characterizes Western Man. The inability to take up and utilize glucose leads to increased use of fatty acids in mitochondrial energy generation, which in turn leads to increased mitochondrial superoxide and hydrogen peroxide generation, which leads to more NF-k B translocation (and you know what follows), more inflammation.
Oxidative stress is damaging to mitochondrial DNA, leading to mitochondrial apoptosis and loss of energy generation (limited exercise capacity, difficulty in weight control). Insulin up regulates eNOS activity; this tonic up regulation in endothelial tone is lost in the setting of insulin insensitivity. Exogenous insulin and insulin sensitizing agents inhibit NOX, up regulate IKBA, and blunt NF-k B translocation, with a secondary reduction in endothelial adhesion molecules, MCP-1, and PAI-1. Conversely, glycated molecules (AGEs, advanced glycation end products) will ligate RAGE (receptor for AGEs) on the mononuclear cell membrane, stimulating NF-k B translocation.
Impaired glucose utilization leads to inappropriate triglyceride synthesis, and this leads to fatty liver. Here, chronic inflammation leads to cholesterol esterification, a fall in free cholesterol, such that SREBP is shunted to the nucleus, to generate more HMG Co-A Reductase and PCSK9. Impaired skeletal muscle glucose uptake and impaired mitochondrial function leads to fatigue. Fatigued people don’t exercise. They eat more, sit around, and put on visceral fat. Visceral fat is hypoperfused and distressed; the sick adipocytes release chemokines that lead to mononuclear infiltration, the generation of inflammatory cytokines, leading to increased lipid synthesis, and more fatty liver and insulin insensitivity. An imbalance between NO and SO within the myocardium compromises calcium flux across the sarcolemma, compromising energy generation, leading to the diastolic and later systolic dysfunction of diabetic cardiomyopathy.
Diabesity, uncommon two generations ago, is rapidly becoming the norm, and is Western Man’s most important cause of avoidable oxidative stress. Conversely, diabetics have the most to gain with antioxidant strategies.
Hypertension is the consequence of long-standing intimal oxidative stress. Insufficient NO generation leads to hypertrophy and proliferation of vascular smooth muscle cells, and "hardening of the arteries". The resultant elevation if shear stress up regulates intimal NOX and XO, leading to more endothelial dysfunction and more oxidative stress.
Oxidative stress shunts the immune response to Th1/Th17 and away from Th2/Treg, not want we want in our patients with atherosclerosis or heart failure.
Oxidative and inflammatory stress, a four-million-year-old response to life (and species) threatening infection, is being chronically and inappropriately activated by the diet, toxic milieu, and life-style of modern man, creating the viscous cycle that drive atherosclerosis and age-related disease in general. Our job as individuals is to avoid these unnatural oxidative burdens. Our job as clinicians is to counsel our patients as to how to do that, and to intervene, at a level appropriate to the patient's age and disease burden, with pharmaceutical and nutraceutical antioxidant interventions.
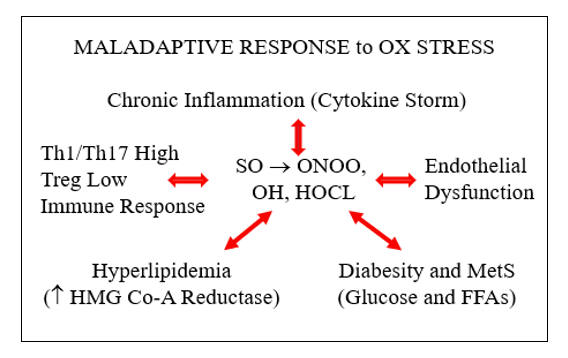
Risk factors are a cause and consequence of oxidative stress. If we lower oxidative stress, atherosclerotic risk factors will improve. If we attenuate one risk factor, oxidative stress and the other risk factors will likewise improve. Our goal as clinicians, stated otherwise, is to replace vicious with virtuous metabolic cycles. The next section discusses strategies to do just that.
The Free Radical Cascade
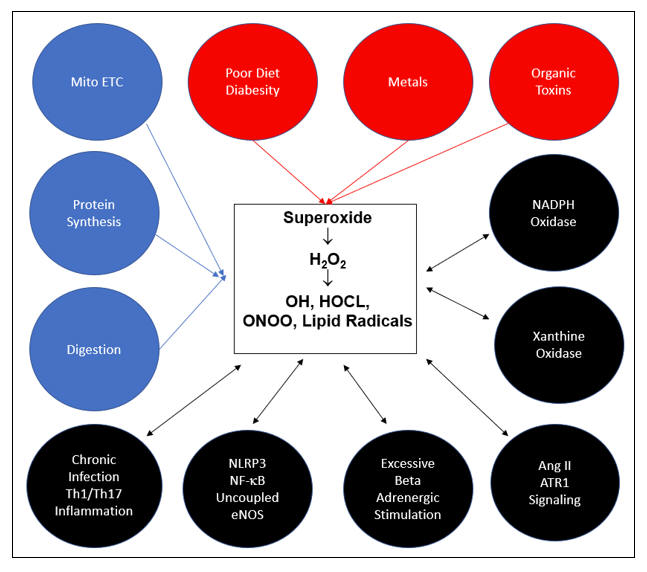
All ROS and RNS descend from superoxide (SO), the sole free radical species that we de novo generate. The above cartoon depicts the pathways of superoxide generation. Constitutive processes, depicted in blue, generate the SO we need to provide the low level H2O2 requisite for cellular signaling. With adequate mineral nutriture, our antioxidant enzymes can detoxify any H2O2 in excess to H2O. In red are the metabolic sins of Modern Man, the key causes of age related acquired oxidative stress. Without supra-normal small molecule antioxidant support, SO and H2O2 overload will occur. ONOO, OH, HOCL, and lipid peroxides will be generated and oxidative distress will follow. As Mother Nature recognizes oxidative stress as evidence of (another) infection, additional superoxide generating systems (NOX, XO, MPO, NLRP3), depicted in black, will be activate, furthering self-inflicted intimal injury. Chronic infection and Th1/Th17 immune response skewing are also causes and consequences of oxidative stress. Vicious cycles are initiated, we get sick, and eventually we die, well before we should.
We don’t want unnecessary sickness, and we certainly do not want
to die before our time. Thus, we need to block these cascades. We can’t let H2O2
"get out of the barn". The common sense approach here is avoid and/or
resolve the three key metabolic sins:
A. Maintain an ideal body weight and take in a clean diet.
B. Organic pollutants generate ROS as our body tries (often without success) to
excrete them. Non-biotransformable pollutants (PCB, Dioxins, and TCDD, the toxin
in Agent Orange) travel within the LDL particle and ligate the Aryl Hydrocarbon
Receptor (the expression of which up regulates in atherosclerosis) within
monocytes. Strategies exist to measure and resolve an organic pollutant burden
(detox supplements, far infrared sauna, and ionic footbath therapy).
C. Toxic metals catalyze free radical chemistry, displace nutritional minerals,
and inhibit our antioxidant enzymes systems. Metal detox is thus of value in the
attenuation of oxidative stress and should have salutary effects in disease
states driven by ROS/RNS overload. The benefit of EDTA-based lead detoxification
in chronic kidney disease has been demonstrated in three separate studies. The
Trial to Assess Chelation Therapy (TACT) demonstrated that EDTA-based metal
detoxification improved outcome in infarct survivors already on standard medical
therapy (which, as we will discuss, blocks SO generating systems), with
particular benefit in diabetics (greater baseline ROS burden).
Patients typically present with active atherosclerosis, the result of decades of ROS-generating diabesity, poor diet, and toxin accumulation. Resolving these factors is important, and in theory could arrest the atherosclerotic process. However, most patients are unable or unwilling to address these issues, and in unstable or symptomatic patients we do not enjoy the luxury of time. Thus, we need to intervene with measures to block ROS-generating enzyme systems, while concomitantly upregulating antioxidant defense capacity.
Before taking aim at oxidative stress, we need to assess its
severity. Thus, our first step will be a metabolic survey. Lab analysis will
tell us where oxidative damage is occurring and will quantitate its severity:
A. Lipid oxidation markers include oxidized LDL, lipid peroxides,
malondialdehyde (MDA),
4-hydroxynonenol (4-HNE), and isoprostanes.
B. Protein oxidation markers include 3-nitrotyrosine and protein carbonyls.
C. DNA oxidation results in the formation of 8-hydroxyguanosine (8-OHdG).
D. Depleted glutathione, Co-Enzyme Q10, Vitamin C, and Vitamin E levels provide
us with a mirror image as to the degree of oxidative stress the patients is
currently experiencing.
E. Endothelial function assessment will give us an intimal redox assessment.
We do not need to measure all of the above markers, but some form of baseline assessment is appropriate (obtain the studies that your preferred lab reports and become comfortable with their analysis). The studies can be repeated periodically to guide our therapeutic efforts. The graphic below relates to a man with recurrent CADz, refractory angina, profound insulin insensitivity, and auto-immune disease. With a comprehensive treatment program his symptoms slowly but steadily improved, in lock step with his oxidative stress markers.

We’ve obtained a baseline measurement of oxidative stress. Now we need to tame it.
Antioxidant supplementation has an important role to play, and will be discussed. However, antioxidant supplementation does not address the causes of oxidative stress, and the supplemented molecules may not reach all sites of concern (circulating LDL oxidation is blunted by Vit E but plaque LDL is unaffected). The allopathic approach to atherosclerosis prevention and treatment is to utilize pharmaceutical agents (and is some cases we can use nutraceuticals) to blunt SO generating systems that are inappropriately up regulated. Let’s discuss these:
NADPH Oxidase (NOX)

NADPH Oxidase (NOX) is Mother Nature’s most powerful superoxide (SO) generator. In infection, phagocytic NOX up regulates appropriately to generate the SO "respiratory burst" needed to kill microbes. Genomic NOX down regulation leads to chronic granulomatous disease, with impaired clearing of microbes, while genomic up regulation of NOX is associated with an increased risk of atherosclerosis (from our perspective, NADPH Oxidase is an obNOXious enzyme). Intimal cells, as well as infiltrating leukocytes, all transcribe NOX. NOX is composed of six subunits. Cytochrome b, a complex of gp91phox and p22phox, is permanently embedded within the cell or organelle membrane. Chemical warfare agents kill via overwhelming oxidative injury. These agents are inherently unstable. Thus, their components are generated at different sites, and brought together only immediately prior to their deployment (an action that our spy satellites can detect). Likewise, Mother Nature does not want the full oxidizing fury of NOX to be released outside of the intended response to infection. Thus, NOX components p47phox, p40phox, and p67phox are sequestered within the cytosol. Stimuli that activate NOX phosphorylate Rac, which then translocates the cytoplasmic components to membrane embedded cytochrome b. Activated NOX transfers electrons from cytoplasmic NADPH to generate SO from O2.
NOX’s job is to protect us from microbial death. As atherosclerosis is a maladaptive response of the innate and acquired immune systems to perceived infection with oxLDL, it is not surprising that risk factors and mediators of atherosclerosis lead to upregulated intimal NOX expression.
Not all NOX activation is pathologic. Low level NOX SO generation likely plays a role in the physiologic (< 100 nM) generation of H2O2 needed for cell signaling, growth, and development. Thus, growth factors and insulin play a role in NOX homeostasis. Mediators of pathologic NOX upregulation, leading to SO generation with subsequent high level (> 250 nM) H2O2 conversion, may activate Rac directly or indirectly via the mechanism of up regulated ATR1 expression.
Rac prenylation, downstream from HMG Co-A Reductase, activates rho kinase, which phosphorylates Rac to activate NOX (thus HMG Co-A Reductase inhibition lowers SO generation). ATR1 ligation by AngII increases NOX transcription (ATR1 transcription is up regulated by prenylation of Rho, also downstream from HMG Co-A Reductase). NOX activation on the basis of oscillatory shear explains the focal initiation of atherosclerosis, and likely plays a role in lesion progression (the tighter the narrowing, the greater the oscillatory stress, so more focal SO and less NO). Aldosterone activates NOX in a manner distinct from ATR1 ligation. Other mediators of oxidative and inflammatory stress, as listed above, may directly or indirectly activate NOX.
NOX generated oxidative stress will activate Xanthine Oxidase, a key ROS generator in diabesity, atherosclerosis, and heart failure, and NLRP3, which mediates plaque activation in relation to "frustrated phagocytosis" of intimal crystalline cholesterol. NOX generated SO will combine with NO to form ONOO, to convert eNOS itself into a SO generator, and will convert to H2O2 at a level sufficient to translocate NF-k B to the nucleus, generating inflammatory mediators and inhibiting rebound activation of Nrf-2.
NOX activation recapitulates our response to infection. This is war! Mother Nature has had four million years to get this right. When Western Man lives incorrectly, we generate an oxidative chemical warfare attack against ourselves.
The best approach to NOX is the best approach to HMG Co-A Reductase and to the other pathways of intimal distress - eat and live the way Mother Nature intended. In our patients, we do not have this luxury of time, and thus pharmaceutical and nutraceutical NOX blockade is appropriate. HMG Co-A Reductase inhibition, ARB, lipophilic ACEI, aldosterone blockade, hydralazine, nebivolol, and colchicine all blunt intimal NOX assembly or activity. Berberine and polyphenols have activity against NOX. Standard antioxidants do not, but assist by scarfing up the ROS/RNS generated by up regulated NOX.
Xanthine Oxidase Inhibition with Allopurinol

Allopurinol inhibits xanthine oxidase (XO), NADPH Oxidase’s partner in intimal oxidative crime. Xanthine oxidoreductase (XDH) degrades end products of purine metabolism. In a reductive environment, XDH functions as a reductase, utilizing NAD+ as a co-factor, generating uric acid and NADH. In an oxidative environment, sulfhydryl groups on XDH are oxidatively modified, converting XDH in to XO. XO utilizes two molecules of oxygen to generate uric acid and superoxide.
In the setting of energy failure (ischemia, heart failure, diabesity, fatty liver) AMP cannot be recycled to ADP and ATP, and degrades to adenosine. Adenosine converts to xanthine and hypoxanthine, the direct substrates of XO. Conditions associated with adenosine excess stimulate transcription of XDH, and conditions associated with oxidative stress convert XDH to XO (recall that XO up regulation in relation to oscillatory shear plays a role in the focal initiation of atherosclerosis). Mother Nature interprets these stimuli as evidence of microbial invasion, and thus XO’s contribution to the ROS/RNS pool is greatly appreciated.
Clinical gout involves a (NLRP3 mediated) innate immune system inflammatory response to the presence of crystalline uric acid within joints. Allopurinol blocks uric acid generation and thus can be used in the prevention of gout. We can use allopurinol to block oxidative stress anywhere in the body, as at these sites XO will be pathologically up regulated.
In stable angina, allopurinol decreases angina frequency and increases treadmill time and time to ST depression. Allopurinol has no effect on HR and BP; rather this agent works by preventing the heart from wasting precious oxygen to generate SO. Thus, allopurinol attenuates symptoms while also favorably effecting the disease process. XO up regulates ten-fold (just like ACE) in acute coronary syndromes (ACS). Irrespective of how ACS is treated, allopurinol begun in the emergency room improves short- and long-term outcome (why not – the driving force here is too much SO). Allopurinol pre-treatment reduces arrythmia and pump dysfunction following on-pump bypass surgery (as will antioxidants, statins, ARBs, and b blockers) by blunting the ischemia-reperfusion pathophysiology inherent in this procedure. Diabesity and Metabolic Syndrome are associated with oxidative stress. ROS/RNS overload are causes and consequences of diabesity and metabolic syndrome. Allopurinol lowers oxidative markers in these settings, and provides a modest glucose lowering effect (remember, H2O2 activates IKK which serine phosphorylates IRS molecules, leading to insulin insensitivity). Endothelial function and carotid intima-media thickness are favorably affected by allopurinol therapy. While uric acid serves an antioxidant function within the circulation, cellular uptake of uric acid leads to mitochondrial oxidative stress. The kidneys retain filtered uric acid, and thus it is not surprising that low dose allopurinol therapy delays disease progression in patients with renal insufficiency with a concomitant reduction in CV event rate.
Allopurinol thus seems to be of universal value in the treatment and prevention of CV and renal disease. The standard dose of allopurinol is 300 mg/day, but in the literature doses up to 900 mg/day have been utilized. There is a dose-related, non-allergic toxicity to allopurinol, characterized by pruritis, rash, malaise, and fever, which will attenuate with a dose reduction. As allopurinol is cleared via the kidneys, pre-existent renal insufficiency, along with small stature and female gender, are associated with an increased sensitivity to this agent.
The indication for allopurinol is to lower oxidative stress, not to lower uric acid. As a general rule, the greater the baseline uric acid level the greater will be the benefit of allopurinol therapy, but elevated uric acid was not a selection requirement in most of the clinical trials demonstrating benefit. For example, if you are experiencing angina on the basis of 95% circumflex narrowing, there is not much arterial surface area on which XO will generate uric acid and SO. Systemic uric acid will not be elevated, but if we blunt inappropriate conversion of oxygen to uric acid and SO at that site, then angina will attenuate.
Allopurinol can be initiated at 100 mg/day, and advanced in 100 mg/day monthly increments, with a reassessment of oxidative stress markers and serum uric acid at the three months point. Increasing allopurinol from 300 mg to 600 mg/day provides a greater incremental improvement in endothelial function than going from nil to 300 mg/day. Thus, if markers of oxidative stress or endothelial tone have not normalized, the dose can be cautiously, and incrementally increased. Patients with pre-existent renal disease have much to gain from allopurinol, but in this population a little allopurinol will go a long way and thus the starting dose will be 50 mg qod, with slow dose advancement. With any hint of toxicity, the dose can be incrementally decreased to a previously well tolerated level.
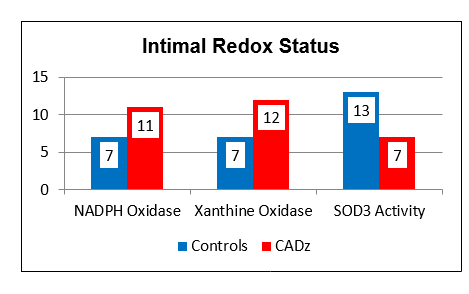
Patients with CADz and CHF demonstrate up regulated NOX and XO
activity. ROS created by NOX converts XDH to XO,
generating more SO, leading to SOD3 depletion.
Intimal Angiotensin II – ATR1 Interaction
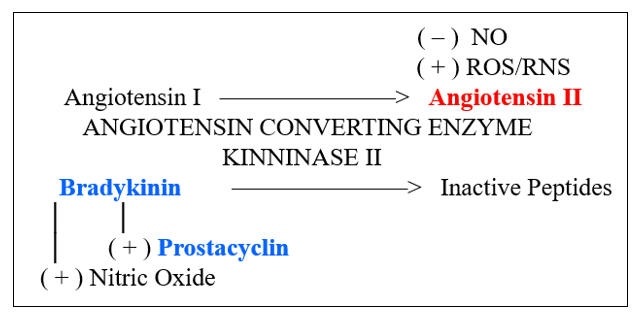
Activation of the RAAS (Renin, Angiotensin, and Aldosterone System) is a primary defense against tissue hypoperfusion. Salt and/or water deficiency were common problems for primitive man. Pathologic vasodilation on the basis of infection threatened to shut down his organ systems. As pathologic upregulation of the RAAS plays a role in hypertension and heart failure, ACEI, ARB, and Aldosterone blockade are recognized as mainstay treatments in these disorders.
From the perspective of atherosclerosis, we need to appreciate that 90% of ACE is intracellular. ACEI expression is inducible, and rises ten-fold in unstable plaques, generating Ang II, which up regulates NOX. While low level ROS generation mediates physiologic growth and differentiation, up regulated NOX and Ang II – ATR1 trafficking leads to vascular smooth muscle proliferation with endothelial intrusion, medial hypertrophy, and left ventricular hypertrophy; stated otherwise, "hardening of the arteries". ANG II – ATR1 signaling translocates NF-k B, which generates Il-6, which stimulates the liver to make more angiotensinogen, creating a pathologic feed forward loop. NF-k B generated COX and PGE2 activation leads to matrix metalloproteinase formation, predisposing to fibrous cap rupture. We need to address this. What processes lead to excess intimal Ang II mediated oxidative stress?
ACE converts Ang I, an inactive precursor, into Ang II, which leads to vasoconstriction, aldosterone generation, and sodium retention. ACE has another role (for which it has another name, kininase II). ACE as kininase II degrades bradykinin into inactive peptides. From the atherosclerosis perspective, bradykinin works in our favor, stimulating eNOS and prostacyclin (to generate NO and vasodilating prostaglandins, respectively).
Standard hydrophilic ACE inhibitors (lisinopril, enalapril, and captopril) work well in hypertension and CHF management. As these agents do not penetrate the intima, they will have no beneficial effect in atherosclerosis (other than lowering excessive intimal shearing stress that would otherwise up regulate NOX and XO). Lipophilic ACEIs (quinapril and ramipril) will work in hypertension and CHF. They will also blunt Ang II generation within the intima, and from the atherosclerosis perspective are clearly preferred.
There is also an intimal RAS, whereby chymase and tryptase convert Ang I into Ang II, an action that is not blocked by lipophilic ACEI. This led to the development of selective Angiotensin Type I Receptor (ATR1) blockade. When Mother Nature opens up a pro-inflammatory, pro-oxidative pathway, she also opens up the door for a counter balancing mechanism. While ligation of ATR1 with Ang II is pro-atherosclerotic, ligation of ATR2 leads to an equal and opposite, endothelial restorative effect. Thus, if we block ATR2 with an ARB (Angiotensin Receptor Blocker, all of which are lipophilic) then Ang II becomes our vasodilating friend (and why combined ACEI and ARB therapy is counterproductive).
Whether lipophilic ACEI or ARB therapy is preferable is a matter of debate. Increased generation of bradykinin leads to the nuisance ACEI cough, and shifts the scale towards ARB therapy. ACEI and ARB therapy may lead to hyperkalemia, and with excessive dosing hypotension and/or renal hypoperfusion. Either agent may produce the uncommon side effect of angioedema, with swelling of the lips and oral mucosa.
As lipophilic ACEI/ARB therapy lowers ROS/RNS generation, you would expect this approach to be effective in the primary and secondary prevention of atherosclerosis. The HOPE study demonstrated a benefit of ramipril in the primary prevention of atherosclerotic events (22% event reduction at two years), not on the basis of BP control (DBP fell only 2 mmHg). An at the time puzzling co-benefit was an attenuation in new onset diabetes and in diabetic consequences. This makes sense to us now, as oxidative stress is both a cause and consequence of DM2. Lipophilic ACEI improves outcome following MI and CABG. Adding lipophilic ACEI or an ARB to a pre-existent statin/aspirin regimen in non-hypertensive, stable atherosclerotic patients did not cause symptomatic hypotension, but did lower circulating levels of ROS, Il-6, TNF-alpha, and endothelial adhesion molecules. We care about inflammation in the circulation, but we care more about what is going on within active plaques. Lipophilic ACEI/ARB therapy added to the pre-operative regimen of individuals scheduled for elective carotid endarterectomy led to a beneficial effect on plaque biochemistry and histology. Plaque lipid content was decreased, with a concomitant increase in smooth muscle and collagen. There were fewer monocytes and T helper cells, less oxLDL and CRP, and lower expression COX, PGE2, and MMP. Bypass surgery leads to profound oxidative stress. We can blunt this with antioxidant therapies such as Vitamin C and Co-Enzymes Q10, but these agents are off limits in the hospital setting. Here we can blunt post-bypass ROS/RNS/ inflammation with statins, ARB/ACEI, and allopurinol, in synergistic fashion.
If risk factors for atherosclerosis are present, then Ang II – ATR1 trafficking is likely up regulated. Ras prenylation downstream from HMG CoA Reductase up regulates AT1R expression. ATR1 stimulation up regulates Rac expression, favoring NOX activation. Sympathetic stimulation leads to renin release, while ATR1 ligation facilitates norepinephrine release from sympathetic nerve endings. As is the case with NF-k B, NO inhibits ATR1 signaling; thus, with endothelial dysfunction ATR1 expression increases. No wonder that Ang II expression is ten-fold up regulated within unstable plaques.
Lipophilic ACEI/ARB intervention makes biologic sense as a first line treatment of hypertension. In patents with known atherosclerosis, unless hypotension is present, blockade of intimal Ang II – ATR1 trafficking assists in the return of the intima to an anti-atherosclerotic reductive environment.
Third Generation b Blockade
|
a 1 |
b 1 |
b 2 |
b 3 |
|
|
HR & Contractility |
|
¯ |
||
|
Vasoconstriction |
|
¯ |
¯ |
|
|
Renin Release |
|
|
||
|
eNOS Activity |
|
|
Catecholamines, norepinephrine and epinephrine, effect change in CV physiology via ligation of adrenergic receptors. Norepinephrine preferentially ligates a 1 and b 1 while epinephrine at physiologic levels interacts only with b receptors.
With respect to oxidative stress and endothelial tone, non-specific b blockade (propranolol or nadolol), via control of hypertension (which increases NOX expression) and inhibition of renin release (which leads to Ang II which increases NOX) will lower intimal oxidative stress. As Th1 lymphocytes bear b receptors, b blockade leads to a desirable shift in immune bias away from Th1 and towards Treg. However, as b 2 ligation mediates smooth muscle relaxation, an undesirable increase in peripheral resistance will occur. b 2 and b 3-mediated eNOS stimulation will be lost, also working against us.
The second generation b 1 specific agents (metoprolol or atenolol) will lower HR, contractility, and renin release, but are less likely to increase afterload, compromise endothelial tone, and precipitate bronchospasm, and were thus a step forward. Third generation agents (carvedilol and nebivolol) provide the standard benefits of b blockade, while adding direct antioxidant protection.
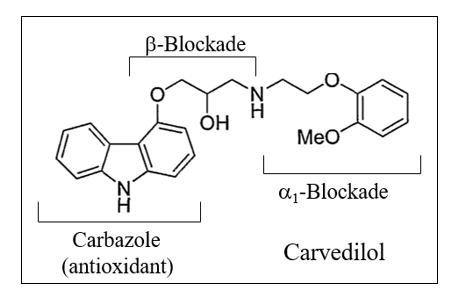
Carvedilol is a non-selective adrenergic blocker. Undesirable inhibition of b 2 mediated vasodilation is neutralized by blockade of norepinephrine mediated a 1vasoconstriction. While loss of b 3-related eNOS up regulation is undesirable, the carbazole side chain of carvedilol provides a counterbalancing antioxidant effect, 10-fold greater than Vitamin E. By squelching ROS, NO loss to ONOO is prevented, and endothelial tone improves.
Oxidative stress is a cause and consequence of heart failure. CHF driving forces (RAAS, catecholamines, inflammatory cytokines, pressure overload) all increase myocardial ROS/RNS generation. Conversely, interventions in our CHF arsenal that improve outcome work, at least in part, by attenuating myocardial oxidative stress. The failing heart is also an overworked heart. The greater the workload, the greater the electron leak from the stressed electron transport chain, the greater the SO generation, the less NADPH available to recharge our backup enzyme-based antioxidant enzymes (GSR, PRX, TRX). Endothelial dysfunction, a sign of intimal oxidative stress, portends a poor outcome in heart failure because SO > NO in the arteries is also SO > NO in the myocardium.
Myocardial ATPase enzymes pump ions across membranes, to coordinate and fuel myocyte contraction and relaxation; all are subject to oxidative inhibition and damage. Sarcolemmal Ca+2-ATPase (SERCA) pumps calcium ions in and out of the sarcoplasmic reticulum. SERCA is inhibited by oxidative stress, as is its transcription. In vitro H2O2-mediated down regulation in SERCA mRNA and protein expression is abrogated by clinically relevant concentrations of carvedilol and N-Acetyl cysteine (both of which "mop up" ROS), but not by metoprolol or propranolol. Furthermore, in the absence of oxidative stress, carvedilol increases SERCA transcription five-fold (carvedilol interacts with the SERCA gene promoter, unrelated to its b -blocker or antioxidant activity).
For these reasons (ROS-scavenging, blockade of a -vasoconstriction, SERCA agonism) carvedilol is preferred in the treatment of heart failure. Circulating oxidative and inflammatory stress markers (oxLDL, MDA, inflammatory cytokines) fall, in concert with improving functional status and ejection fraction. In patients with dilated cardiomyopathy, the addition of carvedilol (mean dose 22 mg/day) to prior therapy with ACEI/ARB, digitalis, and diuretics leads to a 40% reduction in myocardial 4-HNF (a tissue lipid oxidation marker). These myocardial benefits are dose-related. Thus, we start carvedilol at 3.1 mg twice a day, and slowly work up to 25 mg twice a day.
With respect to creating a reductive intimal environment, nebivolol takes us a step further. Nebivolol (5-40 mg/day) is by far the most b 1 specific agent. Thus, loss of b 2 vasodilation and eNOS up regulation is not an issue. Furthermore, nebivolol is a b 3 agonist, stimulating eNOS activity. Nebivolol also directly inhibits NOX, blunting de novo SO formation.
As you would expect, in trials comparing carvedilol and nebivolol to bioequivalent doses (same effect on HR and BP) of metoprolol or atenolol, the third-generation agents provide unique benefits. Lab markers of oxidative stress, such as oxLDL, malondialdehyde, and 8-isoprostanes fall, and endothelial function is more likely to improve. Animal models demonstrate improved intimal and myocardial histology, with attenuated muscle cell hypertrophy, proliferation, and fibrosis. Nuisance side-effects such as fatigue and weight gain are also less likely to occur.
If b blockade is clinically indicated (angina, hypertension, arrhythmia), the third-generation agents will get the job done, with concomitant neutralization of the oxidative distress and impaired endothelial tone that underlie CV disease states.
Colchinine and NLRP3
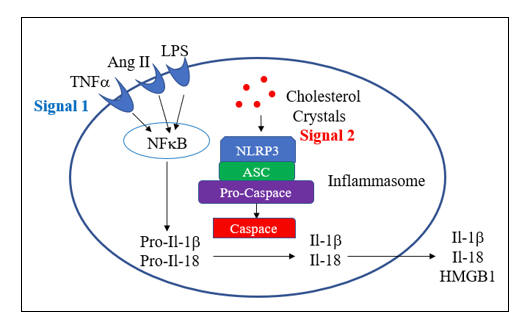
NLRP3, the pathomechanism of acute gout, plays a key role in plaque destabilization. In a sense, you can think of ACS as "gouty arteritis". In gout, uric acid crystals slowly and silently build up within joint tissue. Gouty attacks typically follow an inflammatory incident, such as infection, fever, or a night on the town.
The NLRP3 inflammasome acts as an intra-cytoplasmic mediator of sterile inflammation. Mononuclear cells can phagocytose and degrade microbes, but they can not dissolve rocks. To clear particulate matter (asbestos, silica, and crystalline uric acid and cholesterol) a "flushing" sterile inflammatory response is needed.
Recall that NF-k B transcribes over 100 pro-inflammatory mediators. Interleukin 1b (Il-1b ) and Il-18, are actually transcribed in nascent form. To exert their inflammatory potential, they must first be activated by caspace, which is generated within the activated NLRP3 inflammasome. Activated Il-1b and Il-18 lead to further NF-k B activation (another feed forward loop). NLRP3 also generates a specific mediator of endothelial activation (HMG Box Protein). Polymorphonuclear leukocyte will be pulled in, NOX will activate, and a "respiratory burst" of SO will follow.
With gout, crystals of uric have built up. NLRP3 is activated, but in the absence of NF-k B translocation, there is no nascent Il-1b or Il-8 to activate. Then the fever, or other cause of NF-k B activation, and all of a sudden you have a hot red toe.
The same phenomena is occurring in the coronaries. The majority of lesional cholesterol is cholesterol ester, but a portion exists in crystalline form. NLRP3 activates, silently at first, but then you experience fever, infection, or smoke one too many cigarettes. NF-k B activates, caspace unleashes the inflammatory power of Il-1b and Il-8, HMG Box Protein creates endothelial dysfunction, PMNs are pulled in, and plaque activation occurs.
This process can be attenuated with colchicine. Colchicine blocks tubulin formation, which is needed to assemble both NLRP3 and NOX. As colchicine concentrate 50-fold in leukocytes, doses non-toxic to other organ systems can prevent or ameliorate gout, vascular inflammation, and plaque activation.
Coronary sinus (reflecting coronary artery specific production) Il-1b , Il-8, and Il-6 (the latter reflecting secondary activation of NF-k B) are increased in ACS patients. 1.5 mg of colchicine given one day pre-catheterization reduces coronary sinus Il-1b , Il-8, and Il-6 to levels found in patients with stable angina. Adding colchicine to standard therapy in stable patients will lower CRP. In a study of 500 Australians with known coronary disease, the addition of colchicine 0.6 mg/day to standard therapy (statin, ACEI/ARB, b blocker, and an anti-platelet agent) lowered three year event rate (ACS, CVA, cardiac arrest) by 66% (75% in those who remained on colchicine over the entire study period). In heart failure, colchicine lowers inflammatory markers but does not improve outcome. Colchicine does not prevent restenosis post-PTCA, but it does attenuate neointimal proliferation is diabetics s/p bare-metal stent placement. Colchicine decreases atrial fib recurrence early post-fib ablation, and is of excepted value in dealing with pericarditis and post-cardiotomy inflammation. The standard dose of colchicine is 0.6 mg/day, while in some studies a dose of 1.2 mg/day was used. The key side-effect of colchicine is dose-related diarrhea, which typically resolves with a reduction to every other day or every third day dosing. As colchicine concentrate in leukocytes and has a 2 day half-life, intermittent dosing will still provide persistent protection. One can make a case for colchicine add-on therapy for all patients with significant atherosclerosis, especially when inflammatory mediators, such as CRP, are elevated.
Hydralazine and Nitrate Tolerance
Three decades ago, the direct vasodilator hydralazine was in common use, in BP control and to provide "afterload reduction" in heart failure, typically paired with a "preload reducing" long-acting nitrate. This concept made hemodynamic sense but did not work well in practice. Hydralazine vasodilation led to reflex sympathetic activation, increasing HR and the potential for coronary ischemia. Hydralazine also stimulates renin release, with consequent activation of the RAAS. Long-acting nitrates provide an endogenous NO boost, certainly of short-term benefit, but curiously we had to keep increasing the dose to obtain the desired effect, a phenomena termed "nitrate tolerance". Later it was learned that long-acting nitrate therapy was associated with an increased ischemic event rate, termed "nitrate toxicity". Stated otherwise, long-acting nitrates reduced symptoms but accelerated the disease. We now understand the problem. As mitochondrial aldehyde dehydrogenase converts a long-acting nitrate to NO, NOX is activated, generating SO, which combines with NO to form ONOO, converting eNOS into a SO factory. The 1986 V-Heft study showed no reduction in mortality with hydralazine/nitrate therapy in CHF, and its use was phased out. African American V-Heft subjects, however, did benefit (African Americans experience less RAAS activation vs. Caucasians). This observation led to A-Heft, which randomized African American CHR patients on standard therapy to hydralazine 75 mg tid and isosorbide dinitrate 40 mg tid vs. double placebo. BP fell only 2-3 mmHg, but quality-of-life scores improved and event rate fell by 33%.
Why is Hydralazine working better now? Well, most A-Heft subjects were receiving a B Blocker, to blunt reflex SNS activation, an anti-RAAS agent, and diuretics were on board to blunt vasodilatory induced fluid retention. The key redox benefit is that hydralazine directly scavenges ROS, particularly ONOO, and it inhibits assembly of NOX. Stated otherwise, like nebivolol, hydralazine blunts NOX SO generation and sops up ROS/RNS created by other maladaptively activated enzyme systems. Provided we cover for reflex SNS/RAAS stimulation, we can make good use of hydralazine as an afterload reducing, radical squelching, NOX inhibitor. Hydralazine is initiated at 10 mg qid, and can be increased to 50 mg qid. Drug induced lupus is an uncommon side-effect, and will not show itself for six months (and resolves with drug discontinuation). Reversible hydralazine-induced hepatotoxicity has also been described.
Other direct or indirect NOX inhibitors will ameliorate nitrate tolerance/toxicity, as will small molecule antioxidant supplements. Some, such as N-Acetylcysteine, which supports both glutathione and hydrogen sulfide expression, will synergize with nitrates to attenuate symptoms while also improving outcome.
Taurine, N-Acetylcysteine, and Hydrogen Sulfide (the new NO)?
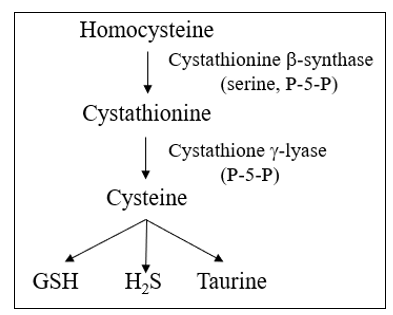
Like NO, hydrogen sulfide (H2S) is enzymatically generated, and
serves as a short-lived physiologic governor. NO nitrosylates, while H2S
sulfhydrates, cysteine moieties on enzymes and signaling molecules (which might
otherwise be oxidized by ROS/RNS). NO and H2S work together to oppose
oxidative stress, and thus protect against CV and other ROS/RNS driven disease
states. H2S up regulates eNOS, and NO protects H2S from
degradation.
Along the gradient from healthy controls to individuals with risk factors, to those with stable vs. unstable angina, we see a progressive decrease in H2S. As H2S appears to be NO’s partner in CV protection, efforts to increase H2S expression are warranted. As outlined above, H2S is derived from homocysteine, within a serine and P-5-P dependent pathway that also produces GSH. Co-synthesized cysteine and taurine also increase production of H2S, helping to explain their across-the-board benefit in CV disease states.
N-Acetylcysteine (NAC) supplementation upregulates GSH expression, and in doses of 500-2000 mg/day has been shown to be of value in hepatic, renal, and pulmonary disease states. NAC ameliorates nitrate toxicity, improves endothelial function, synergizes with ACEI in BP control, and assists in homocysteine clearance (splitting free homocysteine of from its sequestering protein).
Taurine is the most abundantly conserved sulfur-containing amino acid in the
heart. Taurine buffers ions, and thus protects the heart from Ca+2
deficiency or excess, conserves Mg+2, and protects against digitalis
toxicity. Taurine scavenges free radicals, and (unlike Vit E) protects against
HOCL-induced lipid oxidation. Taurine improves endothelial function in smokers
and diabetics, attenuates angina (uncontrolled study), and with or without Co-Q
supplementation relieves symptoms and improves outcome in CHF and in the post-MI
setting. Taurine is helpful in BP control, and in some individuals will assist
in lipid control.
NAC and taurine are inexpensive and non-toxic. In the setting of endothelial dysfunction, known CV disease, or oxidative stress, taurine at 1000 mg bid and NAC at 500 mg bid can do no wrong and a lot of good.
Pentoxifylline to Shift Th1 to Treg
Pentoxifylline (PTX) is not a direct antioxidant. Rather, PTX shifts immune bias away from Th1 and towards Treg, with a resultant reduction in inflammatory cytokine and ROS/RNS generation. PTX has been shown to be of values in Th1 driven states, ranging from psoriasis to renal disease to fatty liver.
Heart failure, irrespective of the cause, is associated with cytokine and/or T cell mediated immune attack against the myocardium. PTX has been shown to attenuated symptoms and/or improve pump function in ischemic, hypertensive, post-partum, and idiopathic cardiomyopathic states. In hypertensive type-2 diabetics, PTX lowers markers of inflammation and increases GSH. PTX improves treadmill time in stable angina, improves outcome in patients undergoing stent placement for ACS, and protects against vein graft failure. PTX is inexpensive and non-toxic. The most common side-effect is dose-related nausea or malaise that typically attenuates over time. A case can be made to utilize PTX, starting at 400 mg daily, and titrating up to 400 mg tid, in patients with CV disease states, particularly if inflammatory makers are elevated.
Berberine
Like pentoxifylline, berberine is not a direct antioxidant. Berberine interacts with Complex 1 of the electron transport chain, creating a biochemical illusion of energy deficiency on the basis of caloric deprivation. As caloric restriction lowers ROS/RNS generation and promotes life extension, the metabolic health benefits of berberine are not surprising.
Berberine activates AMP-sensitive protein kinase (AMPK), which mediates "burn and do not build" signaling. AMPK physiologically down regulates HMG Co-A Reductase, along with the enzymes involved in triglyceride and glycogen synthesis. Berberine protects LDL receptor mRNA from degradation; thus more LDL receptor protein makes it to the hepatocyte membrane. Berberine enters the nucleus and blunts transcription of PCSK9. Thus berberine synergizes with all other means of cholesterol generation inhibition. Berberine increases insulin receptor expression and blunts IKK-mediated serine phosphorylation (and thus down regulation) of IRS proteins, protecting against inflammation-aggravated hyperglycemia. IKK is the same enzyme that shunts NF-k B to the nucleus, and thus we are not surprised that berberine blunts generation of inflammatory cytokines. Berberine promotes translocation of Nrf-2 (which codes for our anti-oxidant and detox enzymes), blunts activation of NOX, improves endothelial tone, and has a favorable effect on the GI microbiome (the first recorded use of berberine was in the treatment of bacterial dysentery).
Berberine is thus of value in the prevention of atherosclerosis, and via its ability to attenuate ROS/RNS/cytokine generation, berberine also helps us deal with its consequences. As in the case of PTX, adding berberine to standard therapy lowers inflammation and improves short-term outcome in ACS patients undergoing PCI. A Chinese study (berberine is a standard therapy in the Orient) demonstrated attenuation of arrythmia, improved pump function, and reduced mortality when berberine was added to standard anti-CHF therapy.
Berberine is inexpensive and non-toxic. Nuisance GI side-effects, primarily constipation or diarrhea, occur in 5-10% of us, and typically resolve with a 50% dose reduction. Berberine itself will not cause hypoglycemia, but if added to insulin or sulfonylurea therapy a reduction in drug dose may be required. The literature includes one case report of berberine-induced bradycardia. The greater the dose or lipid-lowering effect of statin therapy, the greater will be the rise in PCSK9, and thus the greater will be the benefit, and dose-requirement, of add-on berberine therapy. The standard dose of berberine is 500 mg bid; the dose can be advanced to 1000 mg bid, GI tolerance permitting.
Iron Status and Fenton Chemistry
Iron is constitutive in multiple enzymatic steps and is needed for heme synthesis. Iron deficiency is not uncommon and is looked for in routine lab screening. Iron assists in the battle against infection, catalyzing the conversion of H2O2 into microbicidal OH. Primitive man was constantly dealing with infection and red meat was not always available.
H2O2 not neutralized to H2O will be converted, in the presence of ferrous (Fe+2) iron, into OH. OH serves no homeostatic function (other than to kill microbes), and indiscriminately attacks lipids, protein, and DNA. Iron excess will thus lead to intimal oxidative distress, in relation to baseline SO and H2O2 generation, enzymatic antioxidant defense status, and haptoglobin genotype.
Our physiology stores iron as ferritin. Iron excess (ferritin >200 ng/ml) is a risk factor for atherosclerosis, synergizing statistically with a rising LDL. Menstrual cycling limits the potential for iron overload, explaining in part the protective nature of pre-menopausal status (also, estradiol up regulates NO and H2S expression). Iron status relates to dietary iron intake interacting with iron absorptive ability and haptoglobin genotype. Iron deficiency was likely an issue for primitive man, as many of us bear genomic variants coding for heightened iron absorption. Homozygous status for these variants leads to hemochromatosis, with secondary liver, myocardial, and endocrine (oxidative) failure. Heterozygous status or possession of less powerful iron absorption traits leads to the not uncommon finding (12% of us) of iron excess.
In non-anemic individuals with iron excess, periodic blood donation, aiming for a ferritin in the 100s, makes sense. Deferoxamine chelates iron, and can be used when phlebotomy is not possible. The greater the baseline level of oxidative stress, the greater should be the benefit of iron removal. Of interest to our discussion, irrespective of baseline iron status, IV deferoxamine will acutely improve endothelial function in smokers and diabetics.
With respect to atherosclerosis and endothelial tone, iron itself is not the problem, but rather the availability of redox active iron within the intima and in proximity to circulation lipoproteins, which in turn relates to haptoglobin genomic and hemoglobin glycosylation status.
Hemoglobin Glycosylation and Haptoglobin
Genotype
Why Vitamins Don’t (Always) Work
Direct small molecule antioxidant supplementation is an intuitive approach to oxidative stress. If LDL oxidation is the problem, and Vit E (rechargeable with Vit C) serves as a key defense against LDL particle oxidation (Co-Q10 and other lipophilic antioxidants are also involved), then why not supplement?
Vitamin E inhibits LDL oxidation in vitro and in vivo, and is protective in animal models (all wild type for haptoglobin and typically normoglycemic). In industrialized (and now progressively hyperglycemic) man, primary and secondary intervention with Vit E has been shown to be protective in some, but not in all trials. In some studies harm with Vit C is suggested.
These discrepant, and counter intuitive findings can be explained, at least in part, in relation to hemoglobin glycosylation and haptoglobin genomic status.
Total body iron is not the culprit. Rather it is redox active (reduced or ferrous) Fe+2 iron, in proximity to lipid particles or within the intima, that catalyzes pro-atherosclerotic H2O2 to OH conversion. Degenerating rbcs release hemoglobin, exposing the circulation to redox active iron. Haptoglobin, generated by the liver, ligates free hemoglobin and facilitates its clearance (90% hepatic and 10% via mononuclear uptake). Extravascular hemoglobin or heme iron will be cleared by tissue macrophages (which within an active plaque are redox active, churning out their own SO and H2O2).
Haptoglobin exist in two genomic forms, wild type (Hp1) and a variant (Hp2) form, with impaired Hb binding. 16% of us are Hp1/1, 36% Hp1/2, and 48% of us are blessed (from the perspective of iron deficient primitive man battling infection) or cursed (from the perspective of atherosclerotic man) with Hp2/2 status.
Hp2/2 status compromises Hb clearance and iron shielding, as does hemoglobin glycosylation. Diabetics also generate excessive SO and H2O2, and if down regulated for Hp function, then more redox active iron is available to convert these ROS into OH. The poorly cleared Hp2/2-glcoHb complex ligates and oxidizes HDL. HDL thus becomes dysfunctional, with reduced antioxidant and reverse cholesterol transport function.
Given that Hp2/2 status compromises iron clearance, it is not surprising that in men (but not women) iron and ferritin status relate to Hp genomic status.
|
Hp 1/1 |
Hp 1/2 |
Hp 2/2 |
|
|
Serum Iron (umol/l) |
18.6 |
19.2 |
22.6 |
|
Serum Ferritin (ug/l) |
66 |
77 |
128 |
|
Monocyte Ferritin (ug/g) |
326 |
366 |
687 |
Iron, Hp genomic status, and hyperglycemia all interact. Vitamin E will influence the oxidation status of circulating LDL, but does not effect (at least at six weeks) advanced plaque (atherectomy specimen analysis) Vit E or lipid oxidation status. With this understanding of redox biology and Vit E kinetics let’s revisit the Vit E intervention trials.
CHAOS randomized 2002 subjects with newly identified CADz (abnormal angiograms; most with angina or positive stress EKG studies) to placebo vs. Vit E (natural source alpha-tocopherol) 400 IU or 800 IU per day. 8% were diabetic, and in this1996 study pharmaceutical interventions designed to blunt ROS generation were not extensively employed (36% on atenolol and ACEI/statin use was not recorded). 510 day MI risk fell by 77% with Vit E (4.2% vs. 1.4%). The majority of CV deaths occurred early in the study, and here Vit E had no effect.
SPACE randomized 196 hemodialysis patients with pre-existent CV disease (note that dialysis itself generates copious ROS) to Vit E 800 IU (natural source alpha-tocopherol) or placebo. Diabetes prevalence was 43%, and in this 2000 study ACEI therapy was employed in 17%, b blockade in 20%, and 14% wee receiving (type not specified) lipid lowering therapy. At 519 days, an adverse CV event occurred in 33% of placebo subjects vs. 16% in the Vit E group, a 53% risk reduction.
In contrast, Vit E 400 IU per day did not improve outcome in the 2000 HOPE study. 9,541 subjects with stable atherosclerosis (80% coronary, 43% peripheral, and 11% cerebral) or diabetes with one other risk factor (36% of participants) received either placebo or Vit E 400 IU. 39% were on a b blocker and 29% lipid-lowering therapy (none were on ACEI as another arm of HOPE looked at the effect of ramipril, a lipophilic ACEI). 510 day event rate was 16% with Vit E and 15.5% with placebo.
Likewise, the 1999 GISSI study showed no benefit at 3.5 years of Vit E 400 IU per day in 11,324 recent infarct survivors (46% on ACEI, 4% b blockade, and 29% lipid lowering therapy).
In trying to reconcile these studies, we can make two
observations:
A. The greater the level of baseline oxidative stress (not measured but
presumable greater in the CHAOS and SPACE vs. the stable HOPE subjects), the
greater the benefit of Vit E antioxidant intervention.
B. The greater the use of drugs that block ROS generation (statins, ACEI/ARB,
and b blockers), the lower the relative benefit of
Vit E.
Our analysis become more complex, and more informative, if we also consider Hb glycosylation and Hp genomic status. HDL obtained from Hp2/2 diabetics is highly susceptible to in vitro oxidation, a process that is ameliorated by Vit E. Hp1/1 status is protective, especially in diabetics. Hp1/1 status is associated with relative protection against diabetic nephropathy, better outcome post-PCI or post-MI, and better collateral flow. The Strong Heart case-control epidemiology study demonstrated 5-fold and 2.3-fold increased risk for prevalent CV disease amongst diabetics with 2/2 and 1/2 vs. 1/1 Hp status. Amongst non-diabetics, relative risk was less, with 2.3 and 1.4-fold increased risk.
HOPE study data retrospective analysis revealed a greater event rate in placebo-treated diabetic Hp2/2 vs. diabetic Hp1/1 subjects, while Vit E intervention in the diabetic Hp2/2 subjects led to 43% and 55% reductions in MI and CV death. ICARE randomized Hp2/2 diabetic subjects to Vit E 400 IU per day vs. placebo. Hp1/1 and Hp1/2 diabetic subjects were not treated but were monitored for adverse events. ICARE was halted at 18 months, due to the findings of an 2.2% event rate with Vit E vs. 4.7% with placebo. Event rate in diabetic Hp2/2 subjects treated with Vit E was similar to that of diabetic Hp 1/1 and Hp1/2 subjects followed within the ICARE registry. Stated otherwise, Vit E neutralized the pro-oxidant risk associated with Hp2/2 genotype. Looking at the DM/Hp2/Vit E interaction from another angle, post-ICARE subjects coming off Vit E were 4.5 times more likely to experience an adverse during the 18 months post-intervention vs. subjects discontinuing placebo.
Extrapolating these findings to the real world of patient care, it is estimated that administering Vit E to the 42% of diabetics who are homozygous for Hp2 will extend their life three years. Treating 1000 diabetic Hp2/2 diabetics with Vit E over 50 years should prevent 75 MI admits, 31 CABG, and 19 PCI procedures, a risk reduction similar to that of smoking cessation (and superior to that of lifetime statin therapy in high-risk patients and of BP and glucose control in DM2).
How does Hp genotype interact with Vitamin C? The 2004 WAVE study randomized post-menopausal women with abnormal angiograms to bid Vit E 400 IU and Vit C 500 mg vs. bid double placebo. Angiography was repeated at a mean interval of 18 months and change in minimal lumen diameter recorded. Antioxidant supplementation was protective against disease progression in Hp1/1 subjects, particularly in Hp1/1 diabetic subjects. Conversely, there was a trend towards increased disease progression in Hp2/2 subjects, also more pronounced in Hp2/2 diabetics.
Wait a minute? We just said that Hp2/2 diabetics benefit greatly from Vit E. Now WAVE is telling us that Vit E + Vit C intervention may accelerate atherosclerosis in the Hp2/2 population.
While in vitro HDL oxidation in Hp2/2 diabetics is inhibited by Vit E, it is accelerated by Vit C. Vit C reduces iron to its redox active, ferrous status, accelerating conversion of H2O2 to OH. Thus the pro-oxidative effect of Vit C overwhelm the ROS-neutralizing benefit of Vit E (in these in vitro studies, high concentration Vit C was less pro-oxidative, presumably via the mechanism of SO neutralization, such that less H2O2 was available fr conversion in to OH).
A lot of numbers and a lot of biochemistry. How can we use this
understanding of redox biochemistry to reduce risk in our patients? Keeping in
mind that other factors will affect decision making, the following guidelines
make sense:
A. Measure ferritin in all patients. If persistently elevated (ferritin may rise
transiently as an acute phase reactant) take measures to reduce iron burden
(blood donation or iron chelation). This is particularly important in diabetics
and in others with intrinsically increased ROS generation.
B. Genotype for Hp. Vit E supplementation (400 IU per day) makes sense in Hp2/2
individuals, particularly in the setting of DM2. Low dose Vit C mono-therapy may
cause harm in individuals with intrinsically increased ROS generation, elevated
iron stores, or DM2-Hp2/2 status, and is best avoided (at least until these
pro-oxidant phenomena have been addressed).
Optimizing our Innate Enzymatic Antioxidant
Defense
Nrf-2 Translocation, Minerals, GliSODinÔ, and
Pomegranate
Watching the Green Bay Packers defeat the Detroit Lions, my Father told me "remember son, a strong defense wins championships". Father Nature might say "remember Doctor, an optimally expressed mineral-based antioxidant enzyme system is a strong defense against ONOO and OH formation". How can we achieve this strong defense?
Antioxidant enzyme expression, at any given time point, relates to demand and resupply. SOD and Catalase are single-use enzymes. SOD levels are low in CHF and CADz because SO generation is persistently high. GPX and PRX can be recharged by GSR and TRX, but these systems also require periodic resynthesis.
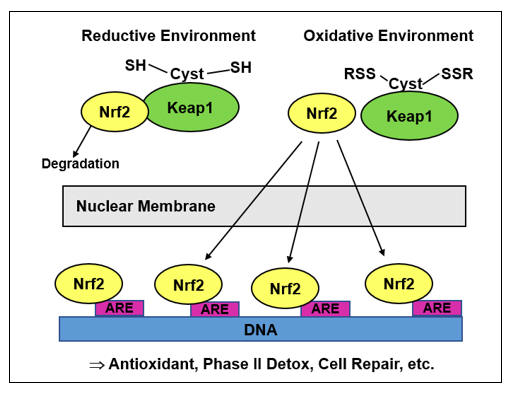
Resupply relates to the extent of Nrf-2 (Nuclear factor-2 erythroid factor-2)
nuclear translocation and ARE (Antioxidant Receptor Element) promoter ligation.
Nrf-2 codes for all antioxidant enzymes, as well as 250 other proteins involved
in cell defense and life protection (phase II detox, cell repair, cell cycle
arrest, apoptosis, and more Nrf-2). Nrf-2 is constitutively transcribed.
Cytoplasmic Nrf-2 is sequestered by Keap1, which promotes its ubiquination and
degradation. The half-life of Nrf-2 is short (in a physiologic, reductive
environment, » 30 minutes). Keap1 contains cysteine
residues with exposed sulfhydryl (SH) groups. SH oxidation by H2O2
leads to Keap1-Nrf-2 dissociation, allowing Nrf-2 to translocate to the nucleus
and ligate ARE promoters. As long as oxidative stress persists, Nrf-2 will
generate antioxidant defense molecules and more Nrf-2. As oxidative stress
resolves (from Mother Nature’s perspective after infection has cleared) Keap1 SH
group oxidation ceases, transcribed Nrf-2 is again sequestered, and antioxidant
enzyme synthesis down regulates.
Primitive Man experienced only intermittent oxidative stress, and here the Nrf-2 system allowed for redox homeostasis. Atherosclerotic Modern Man experiences constant oxidative bombardment (Detroit Lions scenario), and thus stands to benefit from Nrf-2 translocation support.
Exercise improves endothelial function. Recall that pulsatile, laminar flow (exercise or EECP) stimulates eNOS and Nrf-2 transcription. Strenuous exercise also leads to load dependent ROS generation (muscle soreness correlates with ROS-induced muscle damage). Daily runs from Marathon to Sparta could oxidatively shorten your life, but episodic exercise, with longer periods of Nrf-2 rebound (vigorous workouts 2-4 days per week), leads to increased antioxidant/ARE gene expression and health enhancement.
Plants do not wish to be eaten; thus they generate bitter in taste, insecticidal
compounds. Man can derive nourishment from bitter plants as these plant "toxins"
precipitate Nrf-2 translocation, and thus the enzymes needed to neutralize them.
Sulforaphane (found in cruciferous vegetables), curcumin, rosemary, thyme, and
other plant compounds translocate Nrf-2. Some appear to cause a brief, oxidative
stress, altering SH groups on Keap1, while others (berberine and melatonin)
somehow directly enhance Nrf-2 nuclear transport. Nutraceutical firms provide
plant compound blends designed to optimize Nrf-2 trafficking (several can
provide documentation of efficacy).
To prevent oxidative distress, atherosclerosis, and malignancy, a program of regular exercise and dietary plant intake makes sense. In patients with established disease or manifest oxidative stress, Nrf-2 pro-translocation supplementation should lessen SO/H2O2 to ONOO/OH conversion.
Antioxidant (and essentially all other) enzyme systems require mineral co-factors (Mn, Zn, Se, I, Zn, and Cu). It is intuitive that mineral deficiency will compromise antioxidant enzyme status and conversely that repletion will be ameliorative, a position that is supported by epidemiology and the (albeit limited) clinical trial data available.
Pharmaceuticals often deplete minerals, toxic metals antagonize their effects (cadmium displaces zinc and lead and mercury knock out selenium), and the processed Western diet is mineral deficient. Thus mineral repletion makes clinical sense, but which minerals and at what doses? A best answer to this question would require periodic biopsy of our organs with mineral dose titration to optimize enzyme activity in relation to ROS generation. We can’t do this, but as we understand the science we can make sound recommendations.
Acknowledging that excessive dosing of a single mineral can have undesirable consequences (e.g. high dose iodine can adversely affect thyroid function and high dose zinc can deplete copper and vice verse), multi-mineral supplementation is non-toxic and inexpensive and certainly has a role in the prevention and resolution of oxidative stress. Dosing can relate to laboratory assessment of nutrient and ROS status, while a simple approach in the patient with atherosclerosis would be to approximate the mineral content of the TACT-1 (Trial to Assess Chelation Therapy-1) supplement (discussed below).
GliSODinÔ consists of melon-derived SOD, bound to a molecule of gliadin, rendering it absorbable. GliSODinÔ attenuates oxidative stress in vitro and in animal models. In humans, GliSODinÔ (the standard dose is 250 mg bid) protects against HBO-induced oxidative DNA damage and increases SOD expression while reducing CRP in competitive athletes (rowers). In middle aged French men and women with risk factors for atherosclerosis, GliSODinÔ supplementation over three years lowered MDA, increased SOD and GPX expression, and regressed IMT (thus providing a demonstrable anti-atherosclerotic effect). GliSODinÔ is non-toxic (the gliadin moiety appears to be inactive but tolerance in celiac disease has not been specifically studied) and serves as another member of our SO defense squad.
Dietary intake of plant polyphenolic flavonoids, (colorless)
anthoxanthins and (pigmented) anthocyanins (red wine, green and black tea, and
in general a plant based diet), relates inversely to atherosclerosis prevalence
The pomegranate tree is said to have flourished in the Garden of Eden (where
Adam and Eve did well without statins), and its products have been used in the
folk medicine of diverse cultures. Pomegranate (POM) polyphenols neutralize ROS
and (demonstrated in vitro) inhibit NOX and Nf-k B
expression. Relating to this discussion, POM increases transcription and
expression of the paraoxonase (PON) antioxidant enzymes. Intracellular PON2
interacts with Co-Q10 to contain mitochondrial SO. PON1, synthesized
in the liver, associates with HDL within the circulation, using this platform to
excise oxidized lipids within lipoproteins. Stated otherwise, PON1 prevents and
reverses LDL oxidation. Different forms and doses of POM have been utilized in
the (relatively small) intervention trials to date, but these studies have shown
that pomegranate:
A. Increases PON1 expression, lowering (in vivo and in vitro) lipid oxidation in
the circulation.
B. Enters established plaque, there lowering lipid oxidation and preserving GSH.
C. Favorably affects IMT progression (the greater the baseline disease burden,
the more powerful the effect), while decreaasing carotid flow velocities
(suggesting macroscopic disease regression).
D. Attenuates stress induced ischemia (as assessed by stress perfusion imaging).
PON has as second (also POM-enhanced) function, promoting the clearance of
lipophilic organic pollutants and homocysteine thiolactone (homocysteine is
another cause and consequence of oxidative stress).
The best approach to POM supplementation is not certain, but four ounces a day of pomegranate juice is certainly a tasty way to enhance PON expression.
Antioxidant Supplementation
This has become an area of controversy, in that Cardiology journal and book editors cherry pick the studies available and tell us (and the media) that "vitamins don’t work".
We understand that vitamins don’t (always) work, when used in
non-comprehensive fashion, as a drug-like mono-therapy vs. the complicated
ROS/RNS oxidative cascade, and we understand that in certain conditions (iron
overloaded or Hp2/2 diabetics), Vit C mono-therapy could work against us.
However, as practioners of Integrative Medicine (we can use drugs, nutritionals,
interventions, and anything else that might help and won’t hurt), with an
awareness of human physiology, we can make good use of antioxidants, because we
understand that:
A. Antioxidants do not decrease SO generation; they serve only to mitigate the
downstream toxic effects of SO excess.
B. No single vitamin can neutralize all ROS/RNS (Vit E neutralizes lipid
radicals but has no activity against SO or HOCL).
C. No single vitamin is active within all cellular compartments (lipophilic
molecules are inactive in the cytoplasm while Vit C alone can not protect lipid
membranes).
D. Vitamins may not enter all sites of concern (Vit E will blunt LDL oxidation
in the circulation but not within an advanced plaque).
E. Antioxidants work within our physiology as a team (C recharges E and lipoic
acid and bioflavonoids recharge C). Intervening with a single antioxidant is
biologically unreasonable.
Thus, if our patients are to benefit from antioxidant supplementation, it must be comprehensive (mimicking Mother Nature’s designs), and it should supplement (not replace) SO generation and ONOO/OH conversion inhibition strategies. The literature supports this comprehensive approach.
The Antioxidant Supplementation Atherosclerosis Prevention Study (ASAP) looked
at IMT progression in relation to antioxidant supplementation in 510
asymptomatic Finns with cholesterol > 193 mg/dl. Subjects received daily Vitamin
E 272 IU, sustained release Vitamin C 500 mg, both, or placebo. In healthy
Americans, CCA mean-IMT progresses at 0.01 mm/year, while in the Finnish ASAP
population, IMT progression on placebo was 0.02 mm/year. With Vitamin E or
Vitamin C mono-therapy, IMT progression was attenuated by 5%/year, while
combination therapy blunted progression by 35%/year. Combination therapy
provided greater benefit in smokers vs. non-smokers (64% vs. 34% reduction). You
understand why - the greater the baseline level of oxidative stress (DM, CHF,
MI, or smoking as in ASAP), the greater the relative antioxidant need and thus
the greater the upside potential of antioxidant support.
The Indian Experiment of Infarct Survival randomized 125 acute MI patients (all receiving standard therapy) to a 28 day program (IV days 1-3 and then po) of Vits C, E, A and beta carotene vs. placebo. Infarct size (judged by CK release and QRS score) was reduced and 28 day adverse event rate fell from 31% to 21%. Similar results were obtained in a Polish acute MI trial involving a 28 day program of Vit C 2000 mg and Vit E 600 mg, and in post-MI trials involving Co-Q10 with/without other antioxidants.
These studies were carried out 1-2 decades ago, before the wide spread adoption of pharmaceutical ROS inhibition. Will combination antioxidant therapy be of value when added to standard (SO generation suppression) therapy (how we are going to use it)? The best guidance comes from TACT-1 (Trial to Assess Chelation Therapy-1).
| TACT Multi – Six Pills Provides: | |||||||
|
Vit A |
25,000 IU |
Vit B6 |
50 mg |
Magnesium |
500 mg |
Potassium |
99 mg |
|
Vit C |
1,2000 mg |
Folate |
800 mcg |
Zinc |
20 mg |
Inositol |
50 mg |
|
Vit D3 |
100 IU |
Vit B12 |
100 mcg |
Selenium |
200 mcg |
PABA |
50 mg |
|
Vit E |
400 IU |
Biotin |
300 mcg |
Copper |
2 mg |
Boron |
2 mg |
|
Vit K1 |
60 mcg |
Vit B5 |
400 mg |
Manganese |
20 mg |
Vanadium |
39 mcg |
|
Thiamin |
100 mg |
Calcium |
500 mg |
Chromium |
200 mcg |
Citrus Bioflavonoids |
|
|
Niacin |
200 mg |
Iodine |
150 mcg |
Molybdenum |
150 mcg |
100 mg |
|
TACT-1 randomised 1708 infarct survivors on standard therapy (73% statin, 72%
b blocker, 63% ACEI/ARB, and 84% ASA) to 40 weekly
sessions of IV Mg-EDTA (to remove pro-oxidant metals) and/or a three pill bid
broad spectrum antioxidant vitamin and mineral supplement (to attenuate residual
oxidative stress). Metal detoxification led to an 18% reduction in five-year
event rate (38 to 33%). Greater protection was afforded to subjects s/p anterior
as opposed to inferior MI (greater injury and presumably greater subsequent
oxidative stress), with a 37% risk reduction, and in diabetics, in whom event
rate fell by 39%. Antioxidant monotherapy reduced five-year event rate a
non-significant 11% (37 to 34%). Metal detox and antioxidant support
demonstrated event reducing synergy that was not statistically significant, but
the best and worst outcomes were recorded in the double-active and
double-placebo groups, respectively. Synergy was more pronounced in the
diabetics; here double-active therapy reduced five-year death rate by 50%.
Amongst subjects not receiving a statin, antioxidant support decreased event
risk by 38% (36 to 23%).
Special Teams – Mixed Tocopherols and Tocotrienols
|
CHAOS Trial - Non-Fatal MI Rate |
|||
|
Placebo |
Vit E |
Vit E 800 |
Vit E 800 |
|
4.2% |
1.35% |
2% |
0.61% |
In CHAOS (discussed earlier) Vit E (400 or 800 IU/day) decreased 510-day non-fatal MI risk by 77% (4.2% placebo vs. 1.4% Vit E). Vit E works, at least in part, by blunting oxidation of circulating LDL. CHAOS utilized two Vit E dosing levels. Thinking about Vit E the way we think about drugs, more should be better. CHAOS reminds us that nutrients are not drugs; specifically that more a -tocopherol is not better (MI rate 2% with E 800 vs. 0.61% with E 400). How can we explain this?
Tocopherol exists as four homologs (a , b , g , and d ), differing only in orientation and extent of methyl binding at C5 and C7 of its chromanol nucleus. In man, the majority of tocopherol exists as the a -homolog, and thus a decision was made to use pure a -tocopherol in clinical cardiovascular research. The other three homologues exist in nature and seem to play specific roles within our physiology, roles that can not be filled by a -tocopherol at any dose. Specifically, g -tocopherol, but not its a -homologue, can neutralize ONOO, and thus is a good thing to have around.
Tocopherols and tocotrienols share a common, and saturable, GI tract absorption pathway. High dose a -tocopherol will blunt absorption of dietary (and supplemental) b , g , and d -tocopherol and tocotrienols, robbing us of their unique benefits. To get around this problem, mixed tocopherol supplements have been designed, and while not yet subject to clinical trial assessment, when Vit E support is indicated their use makes sense, with or without separate a -tocopherol.

Now the plot speeds up (at least within lipid membranes). In contrast to
tocopherols, tocotrienols contain three double bonds within their lipophilic
side chain. As is the case with saturated vs. unsaturated fatty acids, the
double bonds increase mobility within lipid membranes. Thus tocotrienols provide
40-fold greater lipid radical quenching vs. their less mobile tocopherol
cousins.
Tocopherols blunt lipid LDL oxidation. Tocotrienols blunt LDL oxidation and they blunt LDL generation, via two pathways not related to their antioxidant activity.
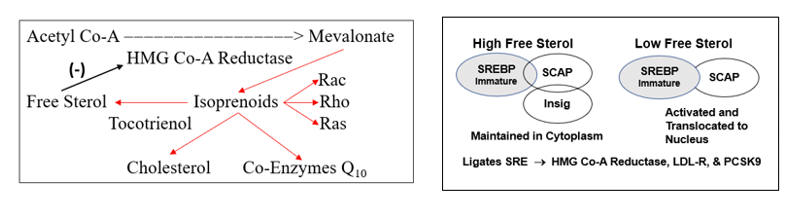
HMG Co-A Reductase (HMG) transcription is under Insig/SCAP free cholesterol sensing control. Post-translational enzyme activity is governed at two additional levels. Berberine and other AMPK agonists promote HMG phosphorylation, which decreases enzyme activity. HMG ubiquination and proteosomal catabolism increases in relation to rising free cholesterol and/or free sterol levels within the cytoplasm. Tocotrienols (but not tocopherols) shunt isoprenoid molecules (which increase NOX and ATR1 and decrease eNOS expression) to their free sterol homologues, thereby accelerating HMG degradation.
Statin drugs and like-acting nutraceuticals are competitive inhibitors of HMG (they look like acetyl-Co-A and gum up enzyme activity). They reduce cytoplasmic free cholesterol and shoot SREBP in to the nucleus. LDL receptor mRNA is transcribed and translated in to membrane LDL receptors, and LDL is cleared from the circulation. The problem here is that SREBP also codes for PCSK9, which degrades the LDL receptor, and for HMG itself. Thus statins set up their own failure (so we keep increasing the dose). We mitigate the PCSK problem with berberine, which blunts PCSK9 transcription. Tocotrienols (but not tocopherols) destabilize HMG mRNA, so less "rebound" HMG is translated.
Clinical trials demonstrate LDL reduction with tocotrienol supplementation, and (as you would expect) synergy with statin agents. Tocotrienols combine LDL reduction with a powerful lipid radical neutralizing effect. Tocotrienols available in supplement form are derived from rice bran, the annatto plant, and palm oil. A 1997 Malaysian study randomized 50 subjects with symptomatic carotid disease to palm oil tocotrienol (240 mg a and g ) or placebo. Plaque burden was stratified as 0-15%, 16-49%, 50-79%, and 80-99%. Over two years, 6/25 placebo subjects progressed one level and 4 by two. Conversely, in the tocotrienol group, only 2 of 25 experienced one level progression; 6 regressed by one level, and 1 subject by two.
This is a small study, but if our goal is to lower LDL and quench lipid peroxides, then tocotrienol supplementation makes sense, particularly in patients who require HMG inhibition/berberine therapy.
Summary and Plan of Action
Atherosclerosis is a maladaptive response of the immune system
to what it perceives as infection of the artery wall with oxidized lipids. To
prevent/attenuate this process we work to achieve physiologic lipid levels and
optimal endothelial tone, with concomitant blunting of intimal lipid trapping,
lipid oxidation, and secondary immune dysregulation. ROS/RNS overload drives
each of these steps. Heart failure, irrespective of etiology, also involves
Th1/Th17-led immune attack against the myocardium and a ROS/RNS-driven
impairment in contractility. Stated otherwise:
SO > NO + Hyperlipidemia = CV Disease
To prevent/resolve oxidative stress, our plan involves:
A. Baseline measures of oxidative stress and its pathologic expression.
1) Lab markers such as oxLDL, MDA, 8-OHdG, GSH, and Co-Q10.
2) Physiologic markers such as endothelial function and carotid IMT.
B. Identify and resolve non-physiologic causes (and consequences) of oxidative
distress:
1) Visceral fat, smoking, poor diet, lack of exercise, sleep apnea, chronic
infection, etc.
2) Insulin insensitivity/DM2.
3) Hypertension.
4) Iron excess (particularly with Hp2/2 genotype).
5) Toxic metals and organic pollutants.
6) Mineral depletion consequent to drug therapy.
C. Pharmaceutical/nutraceutical measures to blunt upregualted
ROS generating systems:
1) HMG Co-A Reductase (drug and non-drug statins + Berberine +Tocotrienols).
2) NADPH Oxidase (HMG inhibition, ACEI/ARB, Spironolactone, Nebivolol,
Hydralazine, Berberine, Colchicine, and Polyphenols).
3) Xanthine Oxidase (Allopurinol)
4) NLRP3 (Colchicine)
5) b -Adrenergic Blockade (Carvedilol and Nebivolol
provide additional antioxidant support)
6) Pentoxifylline to shunt the immune response away from Th1 and towards Treg.
D. Remeasure markers of oxidative stress:
1) If endothelial dysfunction persists intervene with arginine, taurine, and NAC.
2) If oxidative stress persists:
a. Strengthen enzymatic antioxidant defenses with mineral, Nrf-2, GliSODinÔ
, and pomegranate supplementation.
b. Antioxidant supplementation, beginning with a TACT-like multi augmented with
special teams players (taurine to neutralize HOCL and generate HS, NAC to
neutralize ONOO and generate HS, g -tocopherol to
neutralize ONOO, tocotrienols for lipid peroxide neutralization, and Co-Q10 for
mitochondrial support.
E. Repeat baseline measures and apply the above principles until redox status has normalized.
F. Always keep in mind that SO > NO + Hyperlipidemia = CV
Disease
|
Abbreviations |
|||
|
OS |
Oxidative Stress |
Il-6 |
Interkleukin-6 |
|
RNS |
Reactive Nitrogen Species |
TGFB |
Transforming Growth Factor beta |
|
eNOS |
Endothelial Nitric Oxide Synthase |
CAT |
Catalase |
|
NO |
Nitric Oxide |
GPX |
Glutathione Peroxidase |
|
Nrf-2 |
Nuclear factor-2 erythroid factor-2 |
GSR |
Glutathione Reductase |
|
NOX |
NADPH Oxidase |
PRX |
Peroxiredoxin |
|
XO |
Xanthine Oxidase |
TRX |
Thioredoxin |
|
Ang II |
Angiotensin II |
MPO |
Myeloperoxidase |
|
ATR1 |
Ang II Receptor Type 1 |
PON |
Paraoxonase |
|
LPL |
Lipoprotein Lipase |
ADMA |
Asymmetric Dimethylarginine |
|
S-Smase |
Sphingomyelinase |
LIMA |
Left Internal Mammary Artery |
|
PLA2 |
Phospholipase A2 |
SV |
Saphenous Vein |
|
LysoPC |
Lysophosphatidylcholine |
DDAH |
Dimethylaminohydrolase |
|
oxLDL |
Oxidized LDL |
IKK |
Inhibitor of Kappa Beta Kinase |
|
TLR |
Toll-Like Receptor |
IKBa |
Inhibitor of Kappa Beta |
|
MCP-1 |
Monocyte Chemotactic Protein |
LPS |
Lipopolysaccharide |
|
MCSF |
Mono Colony Stimulating Factor |
SRE |
Sterol Regulating Element |
|
NF-k B |
Nuclear Factor Kappa Beta |
LDL-R |
LDL Receptor |
|
AP-1 |
Activator Protein-1 |
Rac |
Activates NADPH |
|
LCAT |
Lecithin Chol Acyl Transferase |
Ras |
Increases ATR1 Expression |
|
PAMP |
Pathogen Activated Molecular Receptor |
||
|
DAMP |
Damage Activated Molecular Receptor |
||
|
APC |
Antigen Presenting Cell |
Rho |
Activated Endothelium |
|
MHC |
Major Histocompatibility Molecule |
AMPK |
AMP Sensitive Protein Kinase |
|
CD4 |
T Helper Cells |
IRS |
Insulin Receptor Substrate |
|
CD8 |
T Surveillance Cells |
HMG |
HMG Co-A Reductase |
|
CD80/86 |
APC Co-Stimulatory Molecules |
AGE |
Advanced Glycation End Product |
|
CD28 |
T Cell CD80/86 Receptor |
RAGE |
Receptor for AGEs |
|
CD28null |
CD4+28 null T Helper Cells are Autonomous and do not require co-stimulation to activate |
||
|
CTLA-4 |
Turns down APC activity |
MDA |
Malondialdehyde |
|
CD40L |
CD40-CD40L APC-T cell co-stimulatory pair |
||
|
NLRP3 |
Nucleotide-binding oligomerization domain-like receptor, pyrin domain-3 inflammasome |
||
|
SO |
Superoxide |
4-HNE |
4-Hydroxynonenol |
|
H2O2 |
Hydrogen Peroxide |
8-OHdG |
8-hydroxyguanosine |
|
ONOO |
Hydroxynitrite |
TNF |
Tumor Necrosis Factor Alpha |
|
OH |
Hydroxyl |
Ifn |
Interferon Gamma |
|
HOCL |
Hypochlorous Acid |
Il-1b |
Interleukin-1 Beta |
|
SCAP |
SREBP Cleavage Activating Protein |
Keap1 |
Kelch-like ECH-associated protein |
|
SREBP |
Sterol Regulating Element Binding Protein |
||
|
PCSK9 |
Proprotein Convertase Subtilisin/Kexin type 9 |
||
|
COX |
Cyclooxygenase |
NAC |
N-Acetylcysteine |
|
PGE2 |
Prostaglandin E2 |
PTX |
Pentoxifylline |
|
MMP |
Matrix Metalloproteinase |
Hp |
Haptoglobin |
|
SERCA |
Sarcolemmal Ca+2-ATPase |
ARE |
Antioxidant Receptor Element |
|
HS |
Hydrogen Sulfide |
POM |
Pomegranate |
References
Atherogenesis
Inflammation and Atherossclerosis. Hansson, G. et. Al. Annu. Rev. Pathol. Mech. Dis. 2006. 1:297-329.
T Cells in Atherogenesis. For Better or For Worse? Robertson, A, and Hansson, G. ATVB. 2006;26:2421-2432.
The role of inflammation, humoral and cell mediated autoimmunity in the pathogenesis of atherosclerosis. Periera, I. and Borba, E. Swiss Med Wkly 2008;138(37-38):534-539.
Role of Monocytes in Atherogenesis. Osterud, Bjarne and BJorklid, Eirik. Physiol. Rev. 83:1069-1112, 2003.
Atherosclerosis – An Inflamatory Disease. Ross, R. NEJM Vol. 340, No. 2, pp. 115-126, 1999.
Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis. Update and Therapeutic Implications. Tabas, I. et. al. Circulation. 2007;116:1832-1844.
Macrophage Foam Cell Formation During Early Atherogenesis Is Determined by the Balance Between Pro-Oxidants and Anti-Oxidants in Arterial Cells and Blood Lipoproteins. Aviram, Michael. Antioxidants & Redox Signaling Vol. 1, No. 4. 1999. pp. 585-594.
The Complex Role of T-Cell–Based Immunity in Atherosclerosis
Aukrust, P. et. al. Current Atherosclerosis Reports 2008, 10:236-243.
Monocyte-Macrophages and T Cells in Atherosclerosis. Tabas, I. and Lichtman, A. Immunity. 2017 Oct. 17;47(4):621-634.
Patients With Acute Coronary Syndrome Show Oligoclonal T-Cell Recruitment With Unstable Plaque Evidence for a Local, Intracoronary Immunologic Mechanism. De Palma, R, et al. Circulation 2006;113:640-646.
Multiple bacteria contribute to intraplaque T-cell activation in atherosclerosis. Van der Meer, JJ, et al. Eur J Clin Invest 2008;38(11):857-862.
Oxidative Stress
Strategies for Reducing or Preventing the Generation of Oxidative Stress. Poljsak, B. Oxidative Medicine and Cellular Longevity Vol. 2011, Article ID 194586.
Oxidative Stress in Human Atherothrombosis: Sources, Markers, and Therapeutic Targets. Martin-Ventura, J. et. al. International Journal of Molecular Sciences 2017, 18, 2315.
Oxidative stress-Mediated Atherosclerosis: Mechansims and Therapies. Yang, X. et. al. Frontiers in Physiology August 2017, Vol. 81, Article 600.
Vascular Oxidative Stress: Impact and Therapeutic Approaches. Sena, C. et. al. Frontiers in Physiology 2018 Dec 4:9:1668.
Oxidative Stress in Atherosclerosis. Kattoor, A. et. al. Curr Atheroscler Rep (2027) 19:42.
ROS Function in Redox Signaling and Oxidative Stress. Schieber, M. and Chanel, N. Curr Biol. 2014 May 19;24(10):R453-R462.
Redox Regulation in the Extracellular Environment. Ottaviano, F. et. al. Circ. J. 2008;72:1-16.
Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Bubici, C. et. al. Oncogene (2006) 25, 6731-6748.
Basic Biology of Oxidative
Stress and the Cardiovascular System. Part 1 JACC 2017;70:196–211.
Impact of Oxidative Stress on the Heart and Vasculature. Part 2. JACC
2017;70:212-29.
Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution. Part 3. JACC 2017;70:230-51.
Endothelial Function and Oxidative Stress
Nitirc Oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Beckman, J. and Koppenol, W. Am. J. Physiol. 271 (Cell Physiol. 40):C1424-C1437, 1996.
Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear sstress. McNally, J. et. al. Am J Physiol Heart Circ. Physiol. 2003 Dec;285(6): H22907.
5-methyltetrahydrofolate rapidly improves endothelial fucntion and dcreases superoxide production in human vessesl: effects on vascualr tetrahydrobiopterin availability and endothelail nitric oxide synthase coupling. Antoniades, C. Circulation. 2006 Sep 12;114(11):1193-201.
NITRIC OXIDE SYNTHASE: Role in the Genesis of Vascular Disease. Cooke, J. and Dzau, V. Annu. Rev. Med 1997 48:489-509.
The Biology and Therapeutic Potential of the DDAH/ADMA Pathway. Arrignoni, F. et. al. Current Pharmaceutical Designs, 2010, 16, 4089-4102.
Role of Endothelial Dysfunction in Atherosclerosis. Davignon, J. and Ganz, P. Circulation 2004;109[suppl III]:III-27-III-32.
Interactions of nitric oxide and peroxynitrite with low-density lipiprotein. Rubbo, H. et. al. Biol Chem 2002 Mar-aprp;383(3):547-52.
Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: Cyclooxygenase-2, managnese sueroxide dismutase, and endothelial cell nitric oxide are selectviely up-regualted by steady laminar shear Stress. Topper, J. et. Al. Proc. Natl. Acad. Sci. Vol. 93, pp. 10417-10422.
Anti-atherogenic effect of laminar shear stress via Nrf2 activation. Takabe, W. et. al. Antioxid Redox Signal 2011 Sep 1;15(5):1415-26.
Does ADMA Cause Endothelial Dysfunction? Cooke, J. Artioscler Thromb Vasc Biol. 2000;20:20132-2037.
Endothelial Function A Critical determinant in Atherosclerosis? Landmesser, U. et. al. Circulation. 2004;109(suppl II):II-27-II33.
Anti-atherogenic effect of laminar shear stress via Nrf2 activation. Takabe, W. et. Al. Antioxid Redox Signal. 2011 Sep 1;15(5):1415-26.
HMG Co-A Reductase
Statins Promote Potent Systemic Antioxidant Effects Through Specific Inflammatory Pathways. Shishebor, M. et. al. Circulation 2003;108:426-431.
Inflammation, Immunity, and HMG-CoA Reductase Inhibitor. Statins as Antiinflammatory Agents? Scheonbeck, U. and Libby, P. Circulation 2004;109[suppl II]:II-18-II-26.
Effects of HMG-CoA Reductase Inhibitors on endothelial Function. Role of Microdomains and Oxidative Stress. Mason, R. et. al. Circulation 2004;109[suppl II]:II-34-II-41.
Potential Role of Statins in Inflammation and Atherosclerosis. Yoshida, M. J Atheroscler Thromb, 2003;10:140-144.
Cellular Antioxidant Effects of Atorvastatin In Vitro and In Vivo. Wassmann, S. et. al. Arterioscler Thromb Vasc Biol. 2002;22:300-305.
Statins stimulate athersoclerosis and heart failure: pharmacological mechanism. Okuyama, H. et. al. Expert Rev Clin Pharmacol. 2015 Mar;8(2):189-99.
Diabesity
Insulin Inhibits Intranuclear Nuclear Factor kB and Stimulates IkB in Mononuclear Cells in Obese Subjects: Evidence for an Anti-inflammatory Effect? Dandona, P. et. al. J Clin Endocrinol Metab 86:3257-3265, 2001.
Nutrients and Oxidative Stress: Friend or Foe. Tan, B. et. al. Oxidative Medicine and Cellular Longevity Vol. 2018, Article ID 971958.
Cardiac Dysfunction and Oxidative Stress in the Metabolic Syndrome: an Update on Antioxidant Therapies. Ilkun, O. and Boudina, S. Curr Pharm Des. 2013;19(27):4806-4817.
Oxidative Stress, Nitric Oxide, and Diabetes. Pitocco, D. et. al. Rev Diabetic Stud (2010) 7:15-25.
Cardiac Dysfunction and Oxidative Stress in the Metabolic Syndrome: an Update on Antioxidnt Therapies. Ilkun, O. and Boudina, S. Curr Pharm Des. 2013;19(27):4806-4817.
NADPH Oxidase
NOX2-dependent regulation of inflammation. Singel, K. and Segal, B. Clin Sci (Lond). 2016 April 01;130(7):479-490.
Vascular Superoxide Production by NAD(P)H Oxidase. Guzik, T. et. al. Circ. Res. 2000 May 12,86(9):E85-90.
Targeting NADPH oxidases in Vascular Pharmacology. Schramm, A. et. al. Vascular Pharmacology 56 (2012) 216-231.
Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear Stress. McNally, J. et. Al. Am J Physiol Heat Circ Physiol 285: H2290-H2297, 2003.
Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. De Keulenaer, G. et. al. Circ. Res. 1998 Jun 1;82(10):1094-101.
Oscillatory Shear Stress Stimulates Endothelial Production of O. from p47phox-dependent NAD(P)H Oxidases, Leding to Monocyte Adhesion. Hwang, J. et. al. The Journal of Biological chemistry. Vol. 278, No. 47, Issue of Nov. 21, pp. 47291-47298, 2003.
Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Selemidis, S. et. al. Cardiovascular research 75 (2007) 349-358.
Vascular Superoxide Production by NAD(P)H Oxidase. Guzik, T. et. al. Circ. Res. 2000;86:e85-e90.
Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. De Keulenaer, G. et. al. Circ. Res. 1998 Jun 1;82(10):1094-101.
Xanthine Reductase and Allopurinol
Imbalance Between Xanthine Oxidase and Nitric Oxide Synthase Signaling Pathway Underlies Mechanoenergetic Uncoupling in the Failing Heart. Saavedra, W. et. al. Circ. Res. 2002;90:297-304.
Elevated Plasma Xanthine Oxidase Activity in Chronic Heart Failure: Sources of Increased Oxygen Radical Load and Effect of Allopurinol in a Placebo Controlled, Double Blinded Treatment Study. Doehner, W. et. al. JACC March 12, 2003 Abstract 1184-81.
Allopurinol Improves Myocardial Efficiency in Patients With Idiopathic Dilated Cardiomyopathy. Cappola, T. et al. Circulation 2001;104:2407-2411.
High-Dose Allopurinol Improves Endothelial Function by Profoundly Reducing Vascular Oxidative Stress and Not by Lowering Uric Acid. George, J. et al. Circulation. 2006;114:2508-2516.
Allopurinol improves endothelial function and reduces oxidant-inflammatory enzyme of myeloperoxidase in metabolic syndrome. Yiginer, O. et al. Clin Res Cardiol 97:334-340 (2008).
Lowering Uric acid With Allopurinal Improves Insulin Resistance and Systemic
Inflammation in Asymptomatic Hyperuricemia. Takir, M. et. Al. J Investig Med
2015;63:924-929.
Effect of high-dose allopurinol on exercise in patients with chronic stable
angina: a randomised, placebo controlled crossover trial. Norman, A. et. al.
Lancet. 2010 Jun 19;375(9732):2161-2167.
The Effects of Allopurinol on the Carotid Intima-Media Thickness in Patients with Type 2 diabetes and Asymptomatic Hyperuricemia: A Three-year Randomized Parallel-controlled Study. Liu, P. et al. Intern Med 54:2129-2137, 2015.
Allopurinol reduces brachial and central blood pressure, and carotid intima-media thickness progression after ischaemic stroke and transient ischaemic attack: a randomised controlled trial. Higgins, P. et. al. Heart 2014;100:1085-1092.
Effects of allopurinol in chronic kidney disease (CKD) progression and cardiovascular risk. Goicoechea, M. et al. Clin J Am Soc. Nephrol. 2010;5:1388-1393.
Allopurinol and Progression of CKD and
Cardiovascular Events:
Long-term Follow-up of a Randomized Clinical Trial. Goicoechea, M. et al. Am J
Kidney Dis. 65(4):543-549.
Allopurinol use and risk of non-fatal acute myocardial infarction. De Abajo, F. et. Al. Heart 2015;101:679-685.
Clinical Study on efficacy of allopurinol in patients with acute coronary syndrome and its functional mechanism. Huang, Y. et al. Hellenic Journal of Cardiology (2017) xx, 1-6 (in press)
The Impact of Allopurinol on Patients With Acute ST Elevation Myocardial Infarction Undergoing Thrombolytic Therapy. Separham, A. et al. J Cardiovasc Pharmacol 2016;68:265-268.
Pretreatment With Antioxidants and Allopurinol Diminishes Cardiac Onset Events in Coronary Artery Bypass Grafting. Sisto, T. et. al. Ann Thorac Surg 1995;59:1519-23.
Renin-Angiotensin-Aldosterone System
Vascular Inflammation and the Renin-Angiotensin System. Braiser, A. et. al. Arterioscler Thromb Vasc Biol. 2002;22:1257-1266.
Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Puja K Mehta and Kathy K. Griendling. Am J Physiol Cell Physiol 292: C82-C97, 2007.
Angiotensin II in inflammation, immunity, and rheumatoid arthritis. Chang, Y. and Wei, W. Clinical and Experimental Immunology 179:137-145. 2014
Blockade of the Angiotensin II Type 1 Receptor Stabilizes Atherosclerotic Plaques in Humans by Inhibiting Prostaglandin E2-Dependent Matrix Metalloproteinase Activity. Cipollone, R, et al. Circulation 2004;109:1482-1488.
Beta Blocker and Angiotensin-Converting Enzyme Inhibitor Therapy is associated with Decreased Th1/Th2 Cytokine Ratios and Inflammatory Cytokine Production in Patients with Chronic Heart Failure. Gage. J. et. al. NeuroImmuneModulation 2004;11:173-180.
Concurrent Treatment With Renin-Angiotensin System Blockers and Acetylsalicylic Acid Reduces Nuclear Factor kB Activation and C-Reactive Protein Expression in Human Carotid Artery Plaques. Sattler, K. et. al. Stroke. 2005;36:14-20.
Irbesartan, an Angiotensin Type 1 Receptor Inhibitor, Regulates the Vascualar Oxidative State in Patients With Coronary Artery Disease. Khan, B. et. al. JACC 2001;38:1662-7.
Angiotensin II cell signaling: physiological and pathological effects in the cardiovascualar system. Mehta, P. and Griendling, K. Am J physiol Cell Physiol 292:C82-C97, 2007.
Effects of an Angiotensin-Converting-Enzyme Inhibitor, Ramipril, on Cardiovascular Events in High-Risk Patientss. NEJM 2000;342:145-153.
b-Adrenergic Blockade
Adrenoreceptors and nitric oxide in the cardiovascular system. Conti, V. et. al. Frontiers in Physiology. Nov. 2013, Vol. 4, Article 311.
Effects of carvedilol on oxidative stress in polymprphonuclear and mononuclear cells in patients with essential hypertension. Am J Med 2004 Apr 1;116(7):460-5.
Protective effects of carvedilol, a vasodilating beta-adrenergic blocker, against in vivo low density lipoprotein oxidation in essential hypertension. Maggi, E. J Cardiovasc Pharmacol. 1996 Apr 27(4):52208.
Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca+2-ATPase 2 gene transcription through modification of Sp1 bindings. Koitabashi, N. et. al. Biochemical and Biophysical Research Communication 328 (2005)116-124.
Novel Mechanisms in the treatment of Heart Failure: Inhibition of Oxygen Radicals and Apoptosis by Carvedilol. Feuerstein, G. et. al. Progress in Cardiovascular Diseases, Vol. 41, No. 1, Suppl. 1 (July/August), 1998: pp. 17-24.
Carvedilol Decreases Elevated Oxidative Stress in Human Failing Myocardium. Nakamura, K. et. al. Circulation. 2002;105:2867-2871.
Antioxidant activity of carvedilol in cardiovascular disease. Dandora, P. et. al. Journal of Hypertension 2007, 25:731-741.
Effects of Carvedilol on Plasma Levels of Pro-Inflammatory cytokines. Kurum, T. et. al. Tex Heart Inst J 2007;34:52-9.
Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. Coats, A. and Jin, S. Journal of Human Hypertension (2017) 31, 376-381.
Nebivolol decreases oxidative stress in essential hypertensive patients and increases nitric oxide reducing its oxidative inactivation. Pasini, A. et. al. Journal of Hypertension 2005, 23:589-596.
Colchinine and NLRP3
Is There a Role for Colchicine in Acute Coronary Syndromes? Nidorf, S. et al. J Am Heart Assoc. 2015;4:e00237.
Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients With an Acute Coronary Syndrome. J Am Heart Assoc. 2015;4e002128.
Effect of colchicine (0.5 mg twice daily) on high-sensitivity C-reactive protein independent of aspirin and atorvastatin in patients with stable coronary artery disease. Nidorf, N. and Thompson, P. Am J Cardiol. 2007 Mar 15;99(6):805-7.
Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. Nidorf, S. et. al. JACC 2013;61:404-10.
Colchicine Treatment for the Prevention of Bare-Metal Stent Restenosis in Diabetic Patients. Deftereos, S. et. al. JACC 2013;61:1679-85.
Colchicine for Prevention of Early Atrial Fibrillation Recurrence After Pulmonary vein Isolation. Deftereos, S. et. al.. JACC 2012;60:1790-6.
Colchicine disposition in human leukocytes after single and multiple oral administration. Chappey, O. et. al. Clin Pharmacol Ther 1993;54:360-7.
NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. Varghese, G. et. al. J Am Heart Assoc. 2016;5e003031 doi;10.1161/JAHA.115.003031.
Coronary Endothelial Dysfunction Induced by Nucleotide Oligomerization Domain-Like Receptor Protein with Pyrin Domain Containing 3 Inflammasome Activation During Hypercholesterolemia: Beyond Inflammation. Zhang, Y. et. al. Antioxid. Redox Signal. 22, 1084-1096.
Interleukin-1b in Coronary Arteries of Patients With Ischemic Heart Disease. Galea, J. et. al. Arteriosclerosis, Thrombosis, and vascular Biology 1996;16:1000-1006.
Molecular mechanism regulating NLRP3 inflammasome activation. Jo, E. et. Al. Cellular & Molecular Immunology (2016) 13, 148-159.
Hydralazine
Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanatin for its beneficial effects on prognosis in patietns with congestive he art failure. Daiber, A. Biochem Biophys Res Commun 2005 Dec 30;338(4):1865-74.B
Nitroso-Redox Balance in the Cardiovascular System. Hare, J. NEJM 351;20 Nov. 11, 2004 pp. 2112-2114.
Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a memebrane-bound NADH oxidase. A new action for an old drug. J Clin Invest 1996 Sep 15;98(6):1465-70.
Combination of Isosorbide Dinitrate and Hydralazine in Blacks with Heart Failure. Tayor, A. et. al. NEJM 2004;351:2049-57.
Impact of Oxidative Stress on the Heart and Vasculature. Munzel, T. et. Al. JACC Vol. 70, No. 2, 2017.
Taurine and N-Acetylcysteine
Supplemental N-acetylcysteine and other measures that boost intracellular glutathione can downregulate interleukin-1b signaling: a potential strategy for preventing cardiovascular events. DiNicolantonio, J. et. al. Open Heart 2017;4e000599.
Taurine protects against low-density lipoprotein-induced endothelial dysfucntion by the DDAH/ADMA patwhay. Tan, B. et. al. Vascul Pharmacol 2007 May;46(5):338-45.
Role of taurine in the vasculature: and overview of experimental and human studies. Abee, W. and Mozaffari, M. Am J Cardiovasc Dis 2011;1(3):293-311.
Boosting endogenous production of vasoprotective hydrogen sulfide via supplementation with taurine and N-acetylcysteine: a novel way to promote cardiovascular health. DiNicolantonio, J. et. al. Open Heart 2017;4:e000600.
The potential health benefits of taurine in cardiovascular disease. Xu, Y. et. al. Exp Clin Cardiol 2008;13(2):57-65.
Comparison of N-acetylcysteine and angiotensin converting enzyme inhibitors in blood pressure regulation in hypertensive patients. Khaledifar, A. et. al. ARYA Atheroscler 2015, Vol. 11, Issue 1.
Pentoxifylline
Pentoxifylline reduces pro-inflammatory and increases anti-inflammatory activity in patients with coronary artery disease-A randomized placebo-controlled study. Fernandes, J, et al. Atherosclerosis 196 (2008) 434-442.
Efficacy of Pentoxifylline in Patients with Stable Angina Pectoris. Insel, J, et al. Angiology – The Journal of Vascular Disease June 1988 pp. 514-519.
Can aortocoronary and peripheral venous bypass graft patency be improved by the administration of Pentoxifylline on a long-term basis? Angelides, N. and Minas, C. Cardiologia 1999;(44):1059-1-64.
Effect of Pentoxifylline on inflammatory burden, oxidative stress, and platelet aggregability in hypertensive type 2 diabetes mellitus patients. Maiti, R., et al. Vascular Pharmacology 47 (2007) 118-24.
Vitamin E and Haptoglobin Genotype
Vitamin E reduces cardiovascular disease in individuals with diabetes mellitus and the haptoglobin 2-2 genotype. Blum S, et. al. Pharmacogenomics. 2010 May;11(5):678-684.
The effect of vitamin E supplementation on cardiovascular risk in diabetic individuals with different haptoglobin phenotypes. Blum, S. et. al. Atherosclerosis. 2010 July;211(1):25-27.
Haptoglobin phenotype is an independent risk factor for cardiovascular disease
in individuals with diabetes: The Strong Heart Study. Levy, A. et. al. J Am
Coll Cardiol 2002;40:1984-90.
Secondary prevention with antioxidants of cardiovascular disease in endstage
renal disease (SPACE): randomised placebo-controlled trial. Boaz, M. et. al.
Lancet 2000;356:1213-18.
Randomized controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Stephens, N. et. al. Lancet 1996;347:781-86.
Vitamin E supplementation and cardiovascular events in high-risk patients (HOPE). Yusuf, S. et. al. N Eng J Med Jan 20;342(3):156-60.
The effect of vitamin therapy on the progression of coronary artery atherosclerosis varies by haptoglobin type in postmenopausal women. Levy, A. et. Al. Diabetes Care 27:925-930, 2004.
The haptoglobin 2-2 phenotype affects serum markers of iron status in healthy
males. Langlois, M. et. al. Clin Chem 46:10 1619-1625 (2000).
Haptoglobin phenotype and coronary artery collateral in diabetic patients
Hochberg, I. et. al. Atherosclerosis 2002 Apr;161(2):441-6.
Vitamin E supplementation reduces cardiovascular events in a subgroup of
middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin
2-2 genotype: a prospective double-blinded clinical trial. Milman, U. et. al.
Arterioscler Thromb Vasc Biol. 2008;28:341-347.
Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet 1999 ;354:447-55.
Vitamin E Supplementation in Patients With Carotid Atherosclerosis. Micheletta, F. et. al. Artioscler Thromb Vasc Biol. 2004;24:136-140.
The effect of supplementation with an antioxidant preparation on LDL-oxidation is determined by haptoglobin polymorphism. Bernard, D. et. al. Redox Report, 8;1, 41-46.
Haptoglobin Genotype Is a Determinant of Iron, Lipid Peroxidation, and Macrophage Accumulaiiotn in the Atherosclerotic Plaque. Arterioscler Thromb Vasc Biol. 2007;27:134-140.
Divergent effects of alpha-tocopherl and vitamin C on the genration of dysfucntional HDL associated with diabetes and the Hp 2-2 genotype. Asleh, R. and Levy, A. Amtioxid. Redox Signal. 12, 209-218.
Intraplaque Hemorrhage. Levy, A. and Moreno, P. Current Molecular Medicine 2006, 6, 6479-488.
Vitamin E Supplementation in Patients With Carotid Atherosclerosis. Micheletta, F. et. al. Arterioscler. Thromb. Vasc. Biol. 2004;24:136-140.
Involvement of Iron-Evoked Oxidative Stress in Smoking Related Endothelail Dysfucntion in Healthy Young Men. Fukami, K. et. al. PLOS ONE Feb. 2014, Vol. 9. Issue 2, pp. 1-16.
Oxidized lipid accumulates in the presence of a-tocopherol in atherosclerosis. Upston, J. et. al. Biochem J. (2002), 753-760.
GlisodinÔ
GLISODINÒ, A VEGETAL SOD WITH GLIADIN, AS PREVENTATIVE AGENT VS. ATHEROSLCEROSIS. AS CONFIRMED WITH CAROTID ULTRASOUND-B IMAGING. Cloarec, M. et. Al. European Annals of Allergy and clinical Immunology. Vol. 39, No. 2, 2007 pp. 45-50.
Effects of Oral Supplementation With Plant Superoxide Dismutase Extract on Selected Redox Parameters and an Inflammatory Marker in a 2,000-m Rowing-Ergometer Test. Skarpanska, A. et. al. International Journal of Sport Nutrition and Exercise Metabolism. 2011, 124-134, 2011.
Supplementation with Gliadin-combined Plant Superoide Dismutase Extract Promotes Antioxidant Defense and Protects Against Oxidative stress. Vouldoukis, I. et. al. Phytother. Res. 18, 957-962 (2004).
Therapeutic value of oral supplementation with melon superoxide dismutase and wheat gliadin combination. Romao, S. Nutrition 31 (2015) 430-436.
Pomegranate and Paraoxonase
Pomegranate Protection against Cardiovascular Diseases. Aviram, Michael and Rosenblat, M. Evidence–Based Complementary and Alternative Medicine. Vol. 2012, Article ID 382763, 20 pages.
Pomegranate juice polyphenols increase recombinant paraoxonase-1 binding to high-density lipiportein: Studies in vitro and in diabetic patients. Fuhrman, B. et. al. Nutritioan 26 (2010) 359-366.
Consumption of Wonderful Variety pomegranate Jucie amd Extract by Diabetic Patients Increases Paraoxonase 1 Association with High-Density Lipoprotein and Stimulates Its Catalytic Activities. Rock, W. et. al. J. Agric. Food Chem. 2008, 56, 8704-8713.
Anti-oxidative effects of pomegranat juice (PJ) consumptoon by diabetic patients on serum and on macrophages. Rosenblat, M. et. al. Athrsoclerosis 187 (2009) 363-371.
Antioxidant and Anti-Inflammatory Role of Paraoxonase 1: Implication in Arteriosclerosis Diseases. Litvinov, D. et. al. N Am J Med Sci. 2012 Nov;4(11):523-532.
Myeloperoxidase, paraoxonase-1, and HDL form a funcitonal ternary complex. Huang, Y. et. Al. J Clin Invest 2013 Sep;123(9):3815-28.
Antioxidant Support
Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: the Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Salonnen, R. et. al. Circualtion 2003 Feb 25; 107(7) 947-53.
Effect of high-dose oral multivitamins and minerals in participants not treated with statins in the randomized Trial to Assess Chelation Therapy (TACT). Issa, O. et. al. Am Heart J. 2018 Jan. 195 70-77.
Antioxidant Vitamins and Their Use in Preventing Cardiovascular Disease. Farbstein, D. et. al. Molecules 2010, 15, 8098-8110.
Serial coronary angiographic evidence that antioxidant vitamin intake reduces priogression of coroanry artery therosclerosis. Hodis, H. et. al. JAMA 1995 Jun 21;273(23):1849-54.
EDTA Chelation Therapy Alone and in Combination with Oral High-Dose Multivitamns and Minerals for Coronary Disease: The Factorial Group Results of the Trial to Assess Chelation Therapy. Lamas, G. et. Al. Am Heart J. 2014 July;168:168(1):37-44.
Oral High-Dose Multivimans and Minerals After Myocardial Infarction. Lamas, G. et. al. Ann Intern Med. 2013;159:797-804.
Nutrients and Oxidative Stress: Friend or Foe? Tan, B. et. al. Oxidative Medicine and Cellular Longevity. Vol. 2018, Article ID 9719584, 24 pages.
Vitamin C and Heart Health: A Review Based on Findings from Epidemiologic Studies. Moser, M. and Chun, O. Int. J. Mol. Sci. 2016, 17, 1328.
Tocotrienols
Biological Properties of Tocotrienols: Evidence in Human studies. Meganathan, P. and Ju-Yen Fu. Int. J. Mol. Sci. 2016, 17, 1682.
Tocotrienol is a cardioprotective agent against ageing-associated cardiovascular disease and its associte dmorbidities. Ramanathan, N. et. Al. Nutr Metab (Lond) 2018 Jan 19;15:6.
Synergistic effect of tocotrienol-rich faction (TRF(25)) of rice bran and lovastatin on lipid parameters in hypercholesterolemic humans. Quereshi, A. et. al. J Nutr Biochem. 2001 Jun 12(6):318-329.
Antioxidant effects of tocotrienols in patietns with hyperlipidemia and carotid stenosis. Tomeo, A. Lipids 1995 Dec;30(12):1179-83.
Palm oil antioxidant effects in patients with hyperlipidemia and carotid stenosis-2 year experience. Kooyenga, D. et. Al. Asia Pac J Nutr. 1997 Mar;6(1):7205.
Melatonin
Mitochondria: Central Organelles for Melatonin’s Antioxidant and Anti-Aging Actions. Reiter, R. Molecules 2018, 23, 509.
Melatonin: An Established Antioxidant Worthy of Use in Clinical Trials. Korkmaz, A. et. al. Mol Med 15(1-2) 43-50, Jan-Feb 2009.
Metals and Oxidative Stress
Oxidative Stress in Lead and Cadmium Toxicity and Its Amelioration. Patra, R. Veterinary Medicine International. Vol. 2011, Article ID 457327, 9 pages.
Lead exposure raises superoxide and hydrogen peroxide in human endothelial and vascualar smooth muscle cells. Ni, Z. et. al. Kidney International, Vol. 66 (2004), pp. 2329-2336.